Article
Addressing problems in gout and hyperuricemia
Gout is a chronic, progressive disease that is managed most effectively when the patient's serum urate level is reduced to lower than 6 mg/dL by early therapeutic intervention.
Gout would seem to be the exemplar of what rheumatologists should aspire to in their understanding and management of rheumatologic disease. The cause-precipitation of sodium urate crystals-is known, and a diagnostic test-recognition of those crystals-is pathognomonic. The metabolic pathways of urate production are well understood, and remarkable progress has been made in acquiring knowledge of the transport processes that are involved in renal elimination. Effective therapeutic agents enable physicians to manipulate both the metabolic and the transport sides of urate balance, to reduce tissue urate concentration and, as a result, to eliminate the supersaturation that leads to crystal formation. The classic recurring episodes of disabling pain usually may be avoided or controlled with use of effective anti-inflammatory agents.1-3


Despite this solid theoretical foundation, problems regularly arise and treatment results often are inconsistent and disappointing. In this article, I consider some recurring concerns in gout management and suggest principles that may help lead physicians to more effective disease control (Table).
1. Complacency
Gout is one of the most painful afflictions of human joints and can be one of the most destructive. However, many patients and physicians consider it a minor inconvenience or even a wry joke. The gouty toe, like the stubbed toe, is seen as a nuisance to be endured with the certainty that a full recovery is not far off. That is not the case.
Gout is a chronic, progressive disease, although the rate of progression varies widely. So long as hyperuricemia persists, the crystals that once may have fired an acute, polymorph-mediated arthritic “flare” will remain in the joint, where they then may feed a more subtle, but far more destructive, macrophage-mediated invasion.4 Then, the effects of these crystals will be amplified silently as they recruit additional crystals, ultimately forming a “urate icing” over articular structures and tophi in juxta-articular tissues. By the time that tophi become apparent on examination or detectable with imaging, far more extensive microscopic deposition is almost certain to have long been in progress.5 This is the process that destroys joints, may lead to major renal impairment, and greatly complicates subsequent efforts to control the hyperuricemia.
Currently, however, there is no method that permits detection of these subtle deposits and gauging of their rate of progression. That rate may be glacially slow at the onset, when recurrent attacks are rare or not apparent. Frequent episodes (2 or 3 in a 1-year period) almost certainly signify progression.
Knowing the baseline serum urate level helps. Modest hyperuricemia (eg, a level below 8 mg/dL) usually reflects a gouty course that moves slowly and may respond to “lifestyle” interventions (eg, reducing calories and alcohol intake and avoiding high-fructose-content fast foods).6,7 Conversely, when acute arthritis occurs in the presence of severe hyperuricemia (eg, a serum urate level above 10 mg/dL), gout almost certainly is progressing.
Assessment of progression is fundamental to the commitment to long-term hypouricemic therapy. A patient who has experienced repeated episodes or has a high urate concentration or both must understand his or her risk and, with disease progression, the decreasing chances of satisfactory control.
A trial of febuxostat studied 760 patients with hyperuricemia and gout.8 Their average serum urate level was 9.8 mg/dL, and the average disease duration was 12 years. One-third of the study patients had renal disease, and many other candidates were rejected because they had serum creatine values greater than 1.5 mg/dL. Tophi were recognized in one-fourth of the patients, and many others had experienced kidney stones.
Each patient who meets these criteria represents a failure of the current standard of care. The major problems that the patients experienced when effective hypouricemic therapy caused repeated gouty flares document the difficulties of delayed intervention. Earlier, more aggressive treatment of hyperuricemic patients with gout should go far to prevent younger patients from reaching this state. Evidence of worsening renal function in these patients also should weigh heavily in therapeutic decision making.
2. Lack of crystal confirmation
Despite the pathognomonic value of urate crystal recognition, many patients never experience a diagnostic arthrocentesis. In part, this reflects arthritis at sites, such as the midfoot, where aspiration is difficult. More often, however, the omission results from physician reluctance to exacerbate patient suffering with an “unnecessary” ordeal.
Aspiration of the acutely painful gouty joint provides the most immediate relief that any physician can offer. The procedure decompresses the distended joint capsule, and the decompression is sustained through the venting needle path.
Knowledge of this pain relief steels physician resolve and patient acceptance. In addition, the procedure permits use of stains and cultures for identifying possible infectious agents and polarizing microscopy for confirming the presence of crystals in the aspirate. Then, the procedure establishes an unequivocal diagnosis before the patient and his physician embark on a therapeutic journey that carries risk in itself and, usually, is lifelong.
There is room for judgment. It remains reasonable to proceed on the basis of the clinical diagnosis alone when the pain is not severe, the patient is known to be hyperuricemic, and identical episodes have come and gone before. When a panel of experts characterized a series of consecutive patients who had gout, about 50% of their new diagnoses were based on recognition of aspirated crystals.9 That percentage seems about right.
The value of crystal identification does not diminish as the disease progresses. Crystal examination is essential in sorting out patients whose gouty arthritis has been misdiagnosed as another rheumatologic disease (eg, long-standing polyarticular gout often is mistaken for rheumatoid arthritis and tophi are interpreted as rheumatoid nodules). Whenever “rheumatoid” disease is atypical (eg, there is little or no rheumatoid factor, joint involvement is asymmetrical, or the patient is unresponsive to methotrexate), the basic diagnosis should be reconsidered. A diagnostic aspiration with a crystal examination becomes a fundamental part of that process.
Bear in mind that osteoarthritis confers an increased risk of gout in any peripheral joint. Therefore, bunions of the great toes and Heberden nodes of the distal interphalangeal joints of the hands indicate sites of special risk of gout. Consideration of possible gouty arthritis is warranted by flaring, episodic inflammation of these sites, or the acute arrival of a painful effusion in a knee that is known to have chronic degenerative disease. Osteoarthritis rarely is a reasonable alternative diagnosis, but often it is a helpful supporting finding in the search for gouty joints. Again, a simple diagnostic aspiration may provide confirmation.
3. Starting allopurinol
for gouty arthritis
To many physicians, a simple train of logic continues to make sense: A. Allopurinol is the most effective drug for gout; B. My patient has gout; and C. I had better start my patient on allopurinol. What could be wrong with that?
The answer is that allopurinol manages hyperuricemia but initially is bad for gouty arthritis. When any drug lowers the serum urate level below 6 mg/dL, the extant deposits of urate crystals begin to be mobilized. As it is freed, the crystal that was relatively inert in its tophus appears to shed its protective coating of plasma proteins10,11; then, it regains its full potential to promote inflammation. When mobilization of additional crystals occurs in the presence of active, crystal-driven arthritis, it simply adds fuel to the fire.
Therefore, the most appealing strategy is to put out the fire by using whichever anti-inflammatory agents are needed, then establish ongoing prophylaxis and, finally, control the hyperuricemia under that prophylactic protection. This way, the fuel can be removed without igniting another fire.12 The task is fraught with danger, and it does not always succeed, but it remains the best available plan.
4. Stopping allopurinol for gouty arthritis
If effective doses of allopurinol release crystals and those crystals then drive arthritis, should the drug be stopped whenever the arthritis flares? Doing so makes sense in theory. In practice, however, many patients with chronic disease go through frequent flares-some less severe than others-and there never seems to be a perfect time.
In this situation, it has seemed most appropriate to begin with a small dose and build gradually to a consistent, effective level while avoiding the yo-yo effect of frequent on-and-off periods. All too often, the drug is stopped for a flare, the physician and patient both conclude that the flare means that allopurinol was not a good choice, and the drug is not resumed. That patient's disease will progress, and any future hypouricemic agent will cause the same complication.
Therefore, a program that emphasizes consistency best serves the long-term interests of most patients. This means staying with the established dose of allopurinol even though that dose has demonstrated its effectiveness by mobilizing enough crystals to induce a flare.
5. Allopurinol dosage fixed
at 300 mg/d
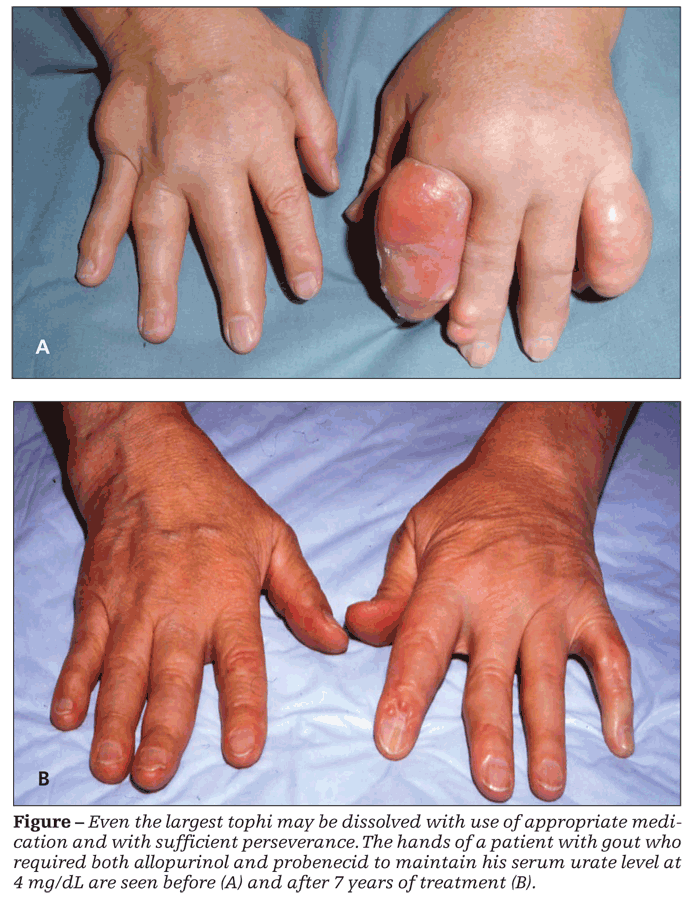
The proper dosage of allopurinol is not 300 mg/d but rather the amount needed to control hyperuricemia. In most patients, this means a target serum concentration of less than 6 mg/dL, although many rheumatologists aim for less than 5 mg/dL when extensive crystalline deposits need to be mobilized. I usually start the dosage at 100 mg/d and increase it by monthly increments of 100 mg/d while monitoring the serum urate level until the necessary dosage has been established. With renal disease or massive tophi (Figure), using 50 mg as both the starting dosage and the amount of each increment may be wisest.


Unless the patient pushes for more rapid control, there is no reason to hurry. Essentially, every patient with gout has been hyperuricemic for many years and no regimen will change his situation overnight. This unhurried approach is taken in the belief that (with concurrent colchicine prophylaxis) it lessens the probability of arthritic flares. That hypothesis has not been confirmed experimentally, however, and it may be found that a more aggressive hypouricemic course (as may become possible with PEGylated uricase preparations) may lead to less overall morbidity.13
In practice, the eventual allopurinol dosage often turns out to be 400 mg/d or more.14 Higher baseline levels and long-standing disease usually imply a higher ultimate dosage; modest hyperuricemia of recent onset is easier to control.
The need to normalize serum urate concentrations applies to patients who have impaired renal function as well as to those whose kidneys are normal. The rare but rightly feared complication of severe hypersensitivity occurs more often when the kidneys are impaired; this complication almost certainly is the result of the renal retention of oxypurinol, the major metabolite of allopurinol.15 Oxypurinol also is the effective agent, however, and renal retention may mean that less drug is required. Often, 200 mg handles the problem well; no patient should receive more than he needs. Also, in theory, no patient should receive less than the needed amount.
A problem occurs when the dosage is increased (occasionally to as much as 800 mg/d) and hyperuricemia persists. This situation may reflect patient noncompliance, which may be tested by measuring the blood oxypurinol level. If no oxypurinol is present, the allopurinol is not being taken. If the oxypurinol concentration is high (eg, greater than 150 µmol/L ), the allopurinol is being taken and there is no present solution, although the addition of probenecid may help control hyperuricemia in patients with normal renal function. When the kidneys are damaged, however, such patients and their physicians now wait in hope that a new agent, such as febuxostat, ultimately will control their hyperuricemia.8
6. Misuse of colchicine (acute)
Darwin might have predicted that over centuries of use, regimens for colchicine would have evolved to a comfortable consensus program that affords maximal benefits with minimal toxicity. That does not seem to have happened. Instead, this valuable but noxious agent has been overused (in the traditional regimen of hourly doses to the point of GI toxicity), compounding the misery of acute gouty arthritis. At the same time, colchicine prophylaxis for gouty flares has been underused and withheld by clinicians who are overly concerned about marrow suppression.
After GI absorption, colchicine moves rapidly into cells, as demonstrated by a 2 L/kg volume of distribution. Because of this intracellular localization, plasma half-lives and concentrations are of little interest to clinicians. Within polymorphonuclear leukocytes, colchicine fights inflammation by inhibiting white cell recruitment to sites of crystallization.16 Conversely, intracellular concentration of colchicine in marrow, nerves, and muscle presumably is responsible for potential toxicity in those organs. Ultimately, both the liver and the kidney participate in elimination of colchicine. Disease in either organ can potentiate its positive and negative properties; therefore, disease in either organ is a reason for caution.17
A useful regimen of colchicine for acute gouty arthritis is an initial 1.2-mg dose followed by another 1.2 mg after 3 to 4 hours and a final 0.6-mg dose at bedtime. Either or both of the latter doses may be lowered or withheld in the event of GI distress or liver disease or if the patient is unusually small. This regimen (adapted from that used widely in France18) is effective when it is used early, although many clinicians prefer to start with NSAIDs (if renal function is normal) or corticosteroids (when the kidneys cause concern). Intra-articular corticosteroid suspensions are the agent of choice for monarticular gouty arthritis in an accessible joint; this is particularly true when the attack is well established (2 or more days), the evolving cascade of inflammation has filled the articular cavity with a “cytokine soup,” and no oral option is likely to result in dramatic improvement.
7. Misuse of colchicine (chronic)
The second widely endorsed application of colchicine is use as prophylaxis against repeated attacks.12 Experience with both allopurinol and febuxostat shows that as little as 0.6 mg/d dramatically reduces this complication of effective hypouricemic therapy.8,19 Higher doses have not been studied as carefully, but they are used frequently, especially in some of the very large men who have gout.
Toxicity may occur in the form of more frequent stools (a feature often welcomed by constipated patients), neuromyopathy, and myelosuppression. Because both the liver and the kidney participate in the clearance of colchicine, concurrent disease in either organ increases the risk of toxicity. These risks also are enhanced by cyclosporine therapy in gouty recipients of transplants.17
All patients should be aware that their physician wants to know about new paresthesias or weakness. Blood cell counts (especially the white blood cell count) should be monitored periodically. However, rheumatologists are experienced in managing drug-induced myelosuppression; perhaps they should consider the example of familial Mediterranean fever, in which colchicine routinely is recommended more aggressively for maximal therapeutic benefit (even in patients who have renal impairment).20
One major concern about colchicine prophylaxis stems from its effectiveness. When the classic attacks of acute gouty arthritis are “swept under the rug” by colchicine suppression, chronic crystal precipitation may progress out of sight and beyond the reach of the examiner's hand. This subtle process may lead to tophi, articular damage, and nephropathy, making treatment far more difficult. Without the warning of sentinel attacks, irremediable injury then takes place while the patient and his physician assume that the problem is under control. Therefore, daily prophylaxis is best reserved for control of drug-induced arthritic flares in patients whose hyperuricemia is being managed with effective hypouricemic therapy.
8. Disease flares during hypouricemic therapy
When probenecid was introduced, it rapidly became apparent that lowering the patient's serum urate concentration leads directly to increasing episodes of gouty arthritis.21 This seeming paradox has been confirmed with the use of each subsequent effective agent, including allopurinol, febuxostat, and PEGylated uricase.8,13,19 Such flares are provoked by the decreasing urate concentration and not by the drug itself. Therefore, the many patients with gout who have stopped effective therapy in the hope that some new hypouricemic agent will avoid this problem almost certainly are destined to wait in vain.
Since the early observations of van Leeuwenhoek, it has been known that gouty tophi are masses of crystals. However, these aggregates rarely cause inflammation. Therefore, it came as a surprise when McCarty and Hollander22 incontrovertibly established that these same crystals of monosodium urate monohydrate induce the dramatic inflammation of classic gouty arthritis.
Subsequent studies from the laboratories of Terkeltaub and of Schumacher explained this dichotomy by showing that the biologically inert crystals of a tophus are coated with an insulating layer of plasma proteins that largely prevents them from activating those cells involved in the acute inflammatory response.10,11 As those crystals begin to dissolve in response to a reduced urate concentration, the protective coating appears to be lost and the mobilized crystals regain their phlogistic potential. It is this “strip mining” of existing crystals, not formation of new ones, that leads to the therapy-induced flare.
Each physician who prescribes therapeutic agents must be aware of this risk and do his utmost to prepare each patient for the real possibility that his gouty arthritis may become transiently worse. Part of this preparation is coprescription of colchicine as a prophylactic agent. The patient also should know that flares may still break through this prophylaxis and, therefore, have a contingency plan, keeping in his mind and in his medicine cabinet the NSAID (with good kidneys) or the corticosteroid that he will use at flare outset. Gouty arthritis becomes less responsive by the hour; patients should not have to wait until they see their physician before they start therapy.
A few patients who have gout-virtually always those with extensive tophi-refuse effective therapy because of recurrent or continuous arthritis or both. Such patients accept progressive, destructive disease because-alone or in combination-the current anti-inflammatory tools of colchicine, NSAIDs, and corticosteroids do not provide adequate control of flares. The existence of these patients should mandate aggressive investigations of new strategies for relief of intractable gouty arthritis. At present, the most promising candidates would seem to be the biologic agents that have now become standard weapons in the armamentarium of every practicing rheumatologist.
9. Disrespect for uricosuric
therapy
Although many patients with gout may attain and maintain target serum urate levels with probenecid,21 this drug rarely is used in the United States. In large part, this reflects an effort to use the simplest possible regimen (once-daily allopurinol rather than twice-daily probenecid) and, therefore, optimize long-term compliance.
In addition, many patients with gout have renal impairment that precludes effective uricosuric therapy. The apparent reason is that probenecid (as well as sulfinpyrazone, benzbromarone, and losartan) is highly protein-bound and therefore bypasses glomerular filtration.23 The drug then must be secreted into the renal tubule before it can begin to inhibit the URAT1 transporter responsible for urate reabsorption. This secretory process is compromised disproportionally by renal impairment; therefore, the uricosuric agent is not effective because it does not reach its intratubular target.24,25
However, some patients-often those who are already taking medications several times a day for concurrent problems-can adhere faithfully to a twice-daily regimen. When such patients have adequate renal function (particularly when they fear the rare problem of severe allopurinol hypersensitivity), use of a dose of probenecid sufficient to keep the serum urate level below 6 mg/dL is perfectly appropriate.
Uricosuric therapy is appropriate for early, intermittent gout. However, allopurinol remains a better choice for patients who have tophaceous gout, renal stones, or a high rate of urinary uric acid excretion.
10. Disregard for renal
problems
Renal impairment continues to figure prominently in the illness of at least half of patients with gout; kidney failure was cited as the cause of death in 20% of patients.26 However, little work has been done in progressive urate nephropathy, and little is known about it. Hypertension and diabetes mellitus both contribute to this complex and multifactorial problem, but neither factor accounts for the medullary crystals of urate that remain demonstrable when autopsy specimens are obtained and preserved in absolute alcohol.27,28 These precipitates are associated with infiltrates of inflammatory cells and seem likely to lead to a significant loss of nephrons.29 Such attrition of nephrons has been recognized increasingly as a cause of hypertension as well as of chronic renal failure.30
Intriguing animal work and epidemiological analysis suggest that additional injury may ensue from hyperuricemia.31 Therefore, both the crystalline solid and the solute in solution may contribute to renal injury. Currently, whether effective control of hyperuricemia will stay or reverse the course of this complication is not known definitively.
Past renal concerns have focused on the recognition and reversal of uric acid overexcretion. With each passing decade, it became more apparent that the true overexcretor is rare. Fewer than 5% of all persons with gout truly overexcrete uric acid, but those who do should be identified to permit proactive handling of their renal risk and to allow for effective genetic counseling and surveillance for family members who may share that risk.
The simplest way to screen for this problem is to measure the rate of uric acid excretion per unit of glomerular filtration rate in a spot urine sample. This useful measurement can be determined by calculating the product of urinary uric acid level and the plasma creatinine level divided by the urinary creatinine level.
References:
References
1. Terkeltaub RA. Clinical practice: gout. N Engl J Med. 2003;349:1647-1655.
2. Choi HK, Mount DB, Reginato AM, et al. Pathogenesis of gout. Ann Intern Med. 2005;143:499-516.
3. Schlesinger N. Management of acute and chronic gouty arthritis: present state-of-the-art. Drugs. 2004;64:2399-2416.
4. Pascual E, Batlle-Gualda E, Martinez A, et al. Synovial fluid analysis for diagnosis of intercritical gout. Ann Intern Med. 1999;131:756-759.
5. Weinberger A, Schumacher HR, Agudelo CA. Urate crystals in asymptomatic metatarsophalangeal joints. Ann Intern Med. 1979;91:56-57.
6. Nakagawa T, Hu H, Zharikov S, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2006;290:F625-F631.
7. Dessein PH, Shipton EA, Stanwix AE, et al. Beneficial effects of weight loss associated with moderate calorie/carbohydrate restriction, and increased proportional intake of protein and unsaturated fat on serum urate and lipoprotein levels in gout: a pilot study. Ann Rheum Dis. 2000;59:539-543.
8. Becker MA, Schumacher HR Jr, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450-2461.
9. Wallace SL, Robinson H, Masi AT, et al. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20:895-900.
10. Terkeltaub RA, Dyer CA, Martin J, Curtiss LK. Apolipoprotein (apo) E inhibits the capacity of monosodium urate crystals to stimulate neutrophils: characterization of intraarticular apo E and demonstration of apo E binding to urate crystals in vivo. J Clin Invest. 1991;87:20-26.
11. Ortiz-Bravo E, Sieck MS, Schumacher HR Jr. Changes in the proteins coating monosodium urate crystals during active and subsiding inflammation: immunogold studies of synovial fluid from patients with gout and of fluid obtained using the rat subcutaneous air pouch model. Arthritis Rheum. 1993;36:1274-1285.
12. Wortmann RL. Effective management of gout: an analogy. Am J Med. 1998;105:513-514.
13. Ganson NJ, Kelly SJ, Scarlett E, et al. Control of hyperuricemia in subjects with refractory gout, and induction of antibody against poly(ethylene glycol) (PEG), in a phase I trial of subcutaneous PEGylated urate oxidase. Arthritis Res Ther. 2006;8:R12.
14. Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, et al. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia: a pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis. 1998;57:545-549.
15. Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity: description and guidelines for prevention in patients with renal insufficiency. Am J Med. 1984;76:47-56.
16. Levy M, Spino M, Read SE. Colchicine: a state-of-the-art review. Pharmacotherapy. 1991;11:196-211.
17. Simkin PA, Gardner GC. Colchicine use in cyclosporine treated transplant recipients: how little is too much? J Rheumatol. 2000;27:1334-1337.
18. Rozenberg S, Lang T, Laatar A, et al. Diversity of opinions on the management of gout in France: a survey of 750 rheumatologists. Rev Rhum Engl Ed. 1996;63:255-261.
19. Borstad GC, Bryant LR, Abel MP, et al. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol. 2004;31:2429-2432.
20. Onen F. Familial Mediterranean fever. Rheumatol Int. 2006;26:489-496.
21. Mason RM. Studies on the effect of probenecid (benemid) in gout. Ann Rheum Dis. 1954;13:120-130.
22. McCarty DJ, Hollander JL. Identification of urate crystals in gouty synovial fluid. Ann Intern Med. 1961;54:452-460.
23. Fanelli GM Jr. Uricosuric agents. Arthritis Rheum. 1975;18:853-858.
24. Guerrini VH, Filippich LJ, English PB, et al. Pharmacokinetics of cefotaxime and probenecid in sheep with normal and reduced renal function. J Vet Pharmacol Ther. 1984;7:283-291.
25. Kamilli I, Gresser U, Pellkofer T, et al. Uricosuric effect of irtemazole in hyperuricemic patients without and with renal insufficiency. Z Rheumatol. 1989;48:307-312.
26. Rosenberg A. Bones, joints and soft tissue tumors. In: Kumar V, Abbas A, Fausto N, eds. Pathologic Basis of Disease. 7th ed. Philadelphia: Elsevier; 2005:1311-1314.
27. Talbott JH, Terplan KL. The kidney in gout. Medicine (Baltimore). 1960;39:405-467.
28. Simkin PA, Bassett JE, Lee QP. Not water, but formalin, dissolves urate crystals in tophaceous tissue samples. J Rheumatol. 1994;21:2320-2321.
29. Sokoloff L. The pathology of gout. Metabolism. 1957;6:230-243.
30. Zandi-Nejad K, Brenner BM. Primary and secondary prevention of chronic kidney disease. J Hypertens. 2005;23:1771-1776.
31. Mazzali M, Hughes J, Kim Y, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension. 2001;38:1101-1106.
















