Article
Behçet syndrome: Taking a systemic approach
Behçet syndrome is a multisystem disease characterized by variable clinical manifestations. The diagnosis is based on recognition of a group of clinical features.
Behet syndrome is a systemic vasculitis of small and large vessels of both the venous and arterial systems the cause of which is unknown.1,2 Hulusi Behet first described Behet syndrome in 1937 in 3 patients who had a triple symptom complex of aphthae, genital ulcers, and hypopyon uveitis.3 Subsequent studies showed that this entity is a multisystem disease characterized by variable clinical manifestations. Virtually all patients have recurring oral aphthae, followed in order of frequency by genital ulcers, skin lesions, arthritis, uveitis, thrombophlebitis, and GI and CNS involvement.
The diagnosis of Behet syndrome is based on recognition of a group of clinical features, because there is no specific symptom or sign. Outcomes have improved over the past 20 years because treatment strategies have been instituted earlier and more aggressively, although vascular involvement and CNS problems are areas that still require better management. Treatment is tailored to the type and severity of symptoms and the patient's sex and age.
In this article, we describe the epidemiology, clinical manifestations, pathology and pathogenesis, and diagnosis and differential diagnosis of Behet syndrome. Then we discuss disease prognosis and management.
EPIDEMIOLOGY
Behet syndrome is most common in the Mediterranean countries and the Far East along the ancient "Silk Route," suggesting that the putative agent or agents, including several genetic factors (eg, HLA-B51), may have spread this way.4 In field surveys carried out in Turkey, the prevalence was between 20 and 421 cases per 100,000 adults5-7; however, there was not a single patient among 47,000 children in another field survey from the same country.8 The disease occurs less frequently in the rest of the world.9-12 Prevalence estimates range from 0.64 cases per 100,000 persons in the United Kingdom to 6.4 cases in Spain to 8.6 cases in the United States.13 However, Turks living in Germany have a very high rate of 77 cases per 100,000 persons; this statistic may point to genetic background rather than environmental elements as the key factor.
Some manifestations show regional differences. GI involvement is common in patients from the Far East14 but rather uncommon in those from Turkey.15
Although a positive pathergy test result is common in patients from Turkey, the Mediterranean countries, and Japan, a positive result is seen less frequently in the Northern European countries and the United States.16 The HLA-B51 association is most pronounced in patients from the Middle East and the Far East.
Disease onset usually occurs in the third decade of life; the condition is rare in older persons (older than 50 years) and younger persons.17 The sexes are affected equally, but the syndrome runs a more severe disease course in men, as well as in younger persons.
CLINICAL MANIFESTATIONS
Mucocutaneous findings
Almost all patients with Behet syndrome have recurring oral ulcerations. These often are the first symptom, and they may precede the other disease manifestations by many years. Minor aphthous ulcers (less than 10 mm in diameter) are the most common type (85%); major or herpetiform ulcers are less common. Although aphthae usually are multiple and occur more frequently in Behet syndrome, they are indistinguishable from those of recurring oral ulcers resulting from other causes.
In men, genital ulcers usually occur on the scrotum and are less common on the shaft or the glans penis; urethritis or dysuria is not a part of Behet syndrome. In women, both the major and minor labia are affected. The ulcers usually heal in 2 to 4 weeks; large ulcers often leave a scar, but small ulcers and those on the minor labia heal without one.18
Skin lesions of various kinds are seen in about 80% of patients with Behet syndrome.19 Acne-like, or papulopustular, lesions are mainly of cosmetic concern. These lesions often are seen not only at the usual acne sites (eg, the face, upper chest, and upper back) but also at unusual sites (eg, the legs and arms). They are indistinguishable from acne vulgaris in both appearance and pathology. 19
Nodular lesions are observed in 50% of patients.20 They usually are confined to the lower limbs in the form of erythema nodosum–like lesions or superficial thrombophlebitis; distinguishing one lesion from the other with the naked eye can be difficult.
The pathergy reaction is a nonspecific hyperreactivity of the skin to trauma, such as a needle prick. A papule or pustule typically forms in 24 to 48 hours after an intradermal injection with a 20-gauge needle. The pathergy positivity is quite specific to patients with Behet syndrome.21 Less common skin concerns include skin ulcers in the axillary and interdigital areas, Sweet syndrome, pyoderma gangrenosum, leukocytoclastic vasculitis, and true arterial lesions.
Eye involvement
Eye disease is seen in half of patients with Behet syndrome but is more common and more severe in men and younger patients.17,22 Eye involvement, which typically occurs within 3 years of disease onset, is a chronic relapsing bilateral uveitis that includes both the anterior and posterior chambers and is a significant cause of morbidity.
Anterior uveitis with intense inflammation (hypopyon) is observed in only a small fraction of patients with eye involvement; typically, it is associated with severe retinal vasculitis. Isolated anterior uveitis occurs infrequently, and conjunctivitis is rare.22 Recurring attacks of eye disease result in structural changes, such as synechiae and retinal scars. If unmanaged, these events eventually lead sto loss of vision.
Musculoskeletal system
Joint disease is observed in half of patients with Behet syndrome in the form of arthritis or arthralgia, each having a similar prevalence.23 Arthritis usually is monarticular or oligarticular but can be symmetrical. Joint disease resolves in a few weeks and seldom leads to deformity and radiological erosions. The knees are involved most often, followed in frequency by the ankles, wrists, and elbows. Back pain is quite rare; controlled studies have not shown increased sacroiliac joint involvement.24
Patients with Behet syndrome who have arthritis have more acne lesions than those who do not.25 In addition, patients with arthritis and acne lesions have significantly higher enthesopathy scores than patients without acne,26 suggesting a link with acne-associated arthritis.
Vascular and cardiac lesions
Vascular involvement, which may include both the venous and arterial systems, is more common in men than in women.27,28 One-third of patients have thrombophlebitis of the deep or the superficial veins, usually in the lower extremities. In rare cases, occlusion of suprahepatic veins can cause Budd-Chiari syndrome, which is associated with a high mortality rate. Although thrombophlebitis occurs very frequently, thromboembolism is rare, most probably because of adherence of thrombi to the diseased veins.
Arterial disease occurs less frequently (in fewer than 5% of cases) but is a serious cause of morbidity and mortality, especially when it involves the pulmonary arteries. Aneurysms may be observed or, less frequently, occlusion of the abdominal aorta or carotid, femoral, popliteal, or coronary arteries.
The mortality rate with pulmonary arterial aneurysms is high,29 especially with an aneurysm less than 3 cm in diameter.30 The main symptom is hemoptysis. Aneurysms may be detected on radiographs, and their presence can be confirmed with CT or MRI.
CNS involvement
Involvement of the CNS, which occurs in 5% to 10% of patients, is one of the most serious manifestations of Behet syndrome, resulting in increased morbidity and mortality.31 Most patients (80%) have parenchymal brain involvement, which mainly affects the brainstem; it is manifested by pyramidal, cerebellar, and sensory symptoms and signs; sphincter disturbances; and behavioral changes. Nonparenchymal disease (20% of cases) takes the form of intracranial hypertension resulting from dural sinus thrombosis and manifested by headaches and papilledema. Simultaneous involvement of the dural sinuses and brain parenchyma is uncommon. The peripheral neuropathy often seen in other vasculitides is rare.
GI involvement
Symptoms include anorexia, vomiting, dyspepsia, diarrhea, and abdominal pain. Mucosal ulceration is seen most frequently in the ileum, followed by the cecum and other parts of the colon.32 Ileocecal ulcers have a distinct tendency to perforate.
Other clinical features
Unlike in many other systemic vasculitides, glomerulonephritis is uncommon in Behet syndrome. AA-type amyloidosis is seen sporadically.33 Epididymitis is seen in about 5% of patients.
PATHOLOGY AND PATHOGENESIS
Although vascular injury is quite common in Behet syndrome and can involve all sizes of vessels in the venous and the arterial systems, frank necrotizing vasculitis of the small and medium-sized arteries of the type seen in antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitides is uncommon. Giant cells and immune complex–type cutaneous venulitis also are uncommon. As a result, vascular involvement in Behet syndrome is rather unique.34
In addition, there is little evidence of vasculitis in some common lesions of Behet syndrome, no evidence of vascular injury in the papulopustular lesions of the skin, and scanty evidence of a frank vasculitis in CNS lesions. Rather unique findings are diffuse inflammatory disease in all layers of the big veins, characteristically involving large segments of the vessel wall; pulmonary arterial aneurysms specific to this condition; and pseudoaneurysms of the big arteries, most probably resulting from vasculitis of the vasa vasorum.
The genetics of Behet syndrome also are uncertain. No clear mendelian pattern emerges. In areas where the disease is endemic, another family member is involved in about 1 of 10 families. The sibling recurrence rate in Turkey has been estimated at 11 to 53 cases per 100,000 persons.35 The most consistent association has been with HLA-B51, which explains only about 20% of the heritability.
There may be some clustering in disease expression in Behet syndrome.36 Through a series of mainly clinical observations, 2 clusters have been described: (1) superficial vein thrombosis, deep venous thrombosis (DVT), and dural sinus thrombi36 and (2) acne, arthritis, and enthesitis.25,26 The association of anti–Saccharomyces cerevisiae antibodies and the presence of inflammatory bowel disease may be another cluster.37 The presence of these clusters may suggest the operation of more than 1 disease mechanism in this complex disorder.
LABORATORY INVESTIGATIONS
There are no specific laboratory findings for Behet syndrome. The erythrocyte sedimentation rate and C-reactive protein level usually are moderately elevated, and they do not correlate well with disease activity.38 Serum immunoglobulin levels sometimes are elevated. Complement levels also may be high. Rheumatoid factor, antinuclear and anticardiolipin antibodies, and ANCA are absent.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS


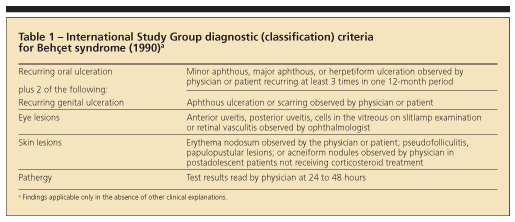
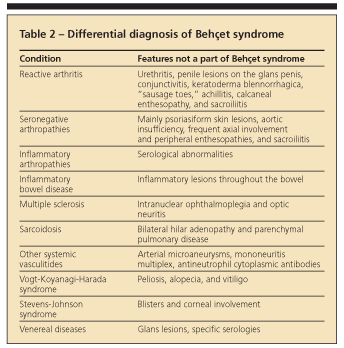
In 1990, the International Study Group published a set of diagnostic (classification) criteria for Behet syndrome. They were based on data obtained by computer analysis of the clinical features of 914 patients with Behet syndrome and 308 controls with disease features that may be confused with those of Behet syndrome.39 These new criteria (sensitivity, 91%; specificity, 96%) have been validated and are used widely (Table 1).40 Recognizing the full-blown syndrome usually is not difficult, but the so-called incomplete forms may present problems (Table 2).


PROGNOSIS AND MANAGEMENT
Many patients with Behet syndrome go into remission with the passage of time.41 Younger patients and men usually have more severe disease-with an increased frequency of mortality and morbidity related to eye, vascular, and neurological disease-than do women and older patients. Loss of useful vision is seen in fewer than 10% to 15% of patients with eye involvement compared with three-fourths of patients 20 to 30 years ago. Although many patients may be managed symptomatically-especially older women-younger men with potentially blinding and lethal disease require aggressive treatment, especially early in the disease course.
Colchicine
This agent is used widely for every lesion of Behet syndrome, but it was useful only for erythema nodosum and arthralgia in a controlled study.42 In a more recent 2-year, placebo-controlled trial, 1 to 2 mg/d of colchicine was beneficial only for genital ulcers, erythema nodosum, and arthritis in women and only for arthritis in men.43
In a 2-year, placebo-controlled trial, the use of azathioprine (AZA), 2.5 mg/kg/d, decreased hypopyon uveitis attacks and the development of new eye disease in patients without eye involvement and preserved visual acuity.44 AZA also was beneficial for oral and genital ulcers and arthritis and, possibly, preventing DVT. However, AZA usually is underdosed and at least 3 months of treatment is required for a beneficial response. Early treatment with AZA also improves patients' long-term prognosis.45
As a rather quick-acting agent, cyclosporine (CYC) usually is the treatment of choice for sight-threatening and progressive uveitis, especially when there is retinal vasculitis46; it is now used at 5 mg/kg/d or at lower dosages because of its adverse effects. Close monitoring is required for hypertension, nephrotoxicity, and neurotoxicity, even at these lower dosages. In resistant cases, CYC often is combined with AZA.
Biologic agents
Success with tumor necrosis factor α (TNF-α) inhibitors (infliximab, then etanercept and adalimumab) has been reported in more than 300 patients with various concerns, such as eye, mucocutaneous, GI, and neurological disease and even pulmonary artery aneurysms. Infliximab has been the most frequently used TNF-α inhibitor in the reports. In a recent position paper, infliximab was recommended as add-on immunosuppressive therapy for select patients with Behet syndrome who are refractory or intolerant to traditional immunosuppressive agents.47 In the only placebo-controlled study of a TNF-α inhibitor in Behet syndrome, etαnercept significantly decreased the mean numbers of oral ulcers and nodular and papulopustular lesions.48
Corticosteroids are used widely for Behet syndrome. In a recent placebo-controlled study, the authors tested the efficacy of depot corticosteroids, 40 mg of methylprednisolone acetate, against placebo in patients with skin-mucosa disease.49 In this trial, depot corticosteroids were useful only in controlling erythema nodosum lesions in women (but not in men).
The European League Against Rheumatism published guidelines for managing Behet syndrome in 2008.50 The recommendations provide good guidance for the management of disease in organ systems other than vascular, neurological, and GI, simply because there is a lack of controlled studies related to the pathologies. Topical measures, including application of local corticosteroids, are sufficient for isolated oral and genital ulcers.
Immunosuppressives, not anticoagulants
Immunosuppressive agents, such as corticosteroids and AZA, are used instead of anticoagulants because the main pathology is inflammation of the vessel wall, and there is a lack of thromboembolism in spite of thrombophlebitis occurring frequently. Cyclophosphamide pulse therapy and corticosteroids are useful for pulmonary and peripheral arterial aneurysms, especially early in the disease course.29 Sulfasalazine, corticosteroids, AZA, TNF-α inhibitors, and thalidomide may be tried for GI disease before surgery. Although corticosteroids are effective for dural sinus thrombosis, managing parenchymal involvement of the CNS is difficult. Corticosteroids, interferon α (IFN-α), AZA, cyclophosphamide, and TNF-α inhibitors may be used.
CONCLUSIONS
More formal, controlled studies are needed for direction in evidence-based treatment of patients with thrombophlebitis and neurological, GI, and arterial involvement. In addition, head-to-head comparisons of TNF-α inhibitors and IFN-α with more traditional drugs (eg, CYC) would be helpful.
References:
References1. Sakane T, Takeno M, Suzuki N, Inaba G. Behçet's disease. N Engl J Med. 1999;341:1284-1291.
2. Yurdakul S, Hamuryudan V, Fresko I, Yazici H. Behçet's syndrome. In: Hochberg MC, Silman AJ, Smolen JS, et al, eds. Rheumatology. 4th ed. Philadelphia: Mosby-Elsevier; 2008:1561-1565.
3. Behçet H. Behcet H. Ãber rezidivierende, aphthose, durch ein Virus verursachte Geshwure am Munde, am Auge und an den Genitalien. Dermatologische Wochenschrift. 1937;36:1152-1157.
4. Verity DH, Marr JE, Ohno S, et al. Behçet's disease, the Silk Road and HLA-B51: historical and geographical perspectives. Tissue Antigens. 1999;54:213-220.
5. Demirhindi O, Yazici H, Binyildiz P, et al. Silivri Fener köyü ve yöresinde Behçet hastaligi sikligi ve bu hastaligin toplum icinde taranabilmesinde kullanabilecek bir yöntem. Cerrahpasa Tip Fak Derg. 1981;12:509-514.
6. Yurdakul S, Günaydin I, Tüzün Y, et al. The prevalence of Behçet's syndrome in a rural area in northern Turkey. J Rheumatol. 1988;15:820-822.
7. Azizlerli G, Köse AA, Sarica R, et al. Prevalence of Behçet's disease in Istanbul, Turkey. Int J Dermatol. 2003;42:803-806.
8. Ozen S, Karaaslan Y, Ozdemir O, et al. Prevalence of juvenile chronic arthritis and familial Mediterranean fever in Turkey: a field study. J Rheumatol. 1998;25:2445-2449.
9. Yamamoto S, Toyokawa H, Matsubara J, et al. A nation-wide survey of Behçet's disease in Japan. Jpn J Ophthalmol. 1974;18:282-290.
10. Jaber L, Milo G, Halpern GJ, et al. Prevalence of Behçet's disease in an Arab community in Israel. Ann Rheum Dis. 2002;61:365-366.
11. Papoutsis NG, Abdel-Naser MB, Altenburg A, et al. Prevalence of Adamantiades-Behçet's disease in Germany and the municipality of Berlin: results of a nationwide survey. Clin Exp Rheumatol. 2006;24(5, suppl 42):S125.
12. Salvarani C, Pipitone N, Catanoso MG, et al. Epidemiology and clinical course of Behçet's disease in the Reggio Emilia area of Northern Italy: a seventeen-year population-based study. Arthritis Rheum. 2007;57:171-178.
13. Calamia KT, Wilson FC, Icen M, et al. Epidemiology and clinical characteristics of Behçet's disease in the US: a population-based study. Arthritis Rheum. 2009;61:600-604.
14. Shimizu T, Ehrlich GE, Inaba G, Hayashi K. Behçet disease (Behçet syndrome). Semin Arthritis Rheum. 1979;8:223-260.
15. Yurdakul S, Tüzüner N, Yurdakul I, et al. Gastrointestinal involvement in Behçet's syndrome: a controlled study. Ann Rheum Dis. 1996;55:208-210.
16. Yazici H, Chamberlain MA, Tüzün Y, et al. A comparative study of the pathergy reaction among Turkish and British patients with Behçet's disease. Ann Rheum Dis. 1984;43:74-75.
17. Yazici H, Tüzün Y, Pazarli H, et al. Influence of age of onset and patient's sex on the prevalence and severity of manifestations of Behçet's syndrome. Ann Rheum Dis. 1984;43:783-789.
18. Mat C, Goksugur N, Engin B, et al. The frequency of scarring after genital ulcers in Behçet's syndrome: a prospective study. Int J Dermatol. 2006;45:554-556.
19. Ergun T, Gürbüz O, Dogusoy G, et al. Histopathologic features of the spontaneous pustular lesions of Behçet's syndrome. Int J Dermatol. 1998;37:194-196.
20. Tursen U, Gurler A, Boyvat A. Evaluation of clinical findings according to sex in 2313 Turkish patients with Behçet's disease. Int J Dermatol. 2003;42:346-351.
21. Tüzün Y, Yazici H, Pazarli H, et al. The usefulness of the nonspecific skin hyperreactivity (the pathergy test) in Behçet's disease in Turkey. Acta Derm Venereol. 1979;59:77-79.
22. Tugal-Tutkun I, Onal S, Altan-Yaycioglu R, et al. Uveitis in Behçet disease: an analysis of 880 patients. Am J Ophthalmol. 2004;138:373-380.
23. Yurdakul S, Yazici H, Tüzün Y, et al. The arthritis of Behçet's disease: a prospective study. Ann Rheum Dis. 1983;42:505-515.
24. Yazici H, Tuzlaci M, Yurdakul S. A controlled survey of sacroiliitis in Behçet's disease. Ann Rheum Dis. 1981;40:558-559.
25. Diri E, Mat C, Hamuryudan V, et al. Papulopustular skin lesions are seen more frequently in patients with Behçet's syndrome who have arthritis: a controlled and masked study. Ann Rheum Dis. 2001;60:1074-1076.
26. Hatemi G, Fresko I, Tascilar K, Yazici H. Increased enthesopathy among Behçet's syndrome patients with acne and arthritis: an ultrasonography study. Arthritis Rheum. 2008;58:1539-1545.
27. Müftüoglu AU, Yurdakul S, Yazici H, et al. Vascular involvement in Behçet's disease: a review of 129 cases. In: Lehner T, Barnes CG, eds. Recent Advances in Behçet's Disease. London: Royal Society of Medicine Services Ltd; 1986:255-260.
28. Lie JT. Vascular involvement in Behçet's disease: arterial and venous and vessels of all sizes. J Rheumatol. 1992;19:341-343.
29. Hamuryudan V, Yurdakul S, Moral F, et al. Pulmonary arterial aneurysms in Behçet's syndrome: a report of 24 cases. Br J Rheumatol. 1994;33:48-51.
30. Seyahi E, Melikoglu M, Akman C, et al. Pulmonary vascular involvement in Behçet's syndrome (BS). Arthritis Rheum. 2007;56(suppl). Abstract 853.
31. Akman-Demir G, Serdaroglu P, Tasçi B. Clinical patterns of neurological involvement in Behçet's disease: evaluation of 200 patients. The Neuro-Behçet Study Group. Brain. 1999;122:2171-2182.
32. Korman U, Cantasdemir M, Kurugoglu S, et al. Enteroclysis findings of intestinal Behçet disease: a comparative study with Crohn disease. Abdom Imaging. 2003;28:308-312.
33. Melikoglu M, Altiparmak MR, Fresko I, et al. A reappraisal of amyloidosis in Behçet's syndrome. Rheumatology (Oxford). 2001;40:212-215.
34. Melikoglu M, Kural-Seyahi E, Tascilar K, Yazici H. The unique features of vasculitis in Behçet's syndrome. Clin Rev Allergy Immunol. 2008;35:40-46.
35. Gül A, Inanç M, Ocal L, et al. Familial aggregation of Behçet's disease in Turkey. Ann Rheum Dis. 2000;59:622-625.
36. Tunc R, Saip S, Siva A, Yazici H. Cerebral venous thrombosis is associated with major vessel disease in Behçet's syndrome. Ann Rheum Dis. 2004;63:1693-1694.
37. Fresko I, Ugurlu S, Ozbakir F, et al. Anti-Saccharomyces cerevisiae antibodies (ASCA) in Behçet's syndrome. Clin Exp Rheumatol. 2005;23(4, suppl 38):S67-S70.
38. Müftüoglu AU, Yazici H, Yurdakul S, et al. Behçet's disease: relation of serum C-reactive protein and erythrocyte sedimentation rates to disease activity. Int J Dermatol. 1986;25:235-239.
39. International Study Group for Behçet's Disease. Criteria for diagnosis of Behçet's disease. Lancet. 1990;335:1078-1080.
40. O'Neill TW, Rigby AS, Silman AJ, Barnes C. Validation of the International Study Group criteria for Behçet's disease. Br J Rheumatol. 1994;33:115-117.
41. Kural-Seyahi E, Fresko I, Seyahi N, et al. The long-term mortality and morbidity of Behçet syndrome: a 2-decade outcome survey of 387 patients followed at a dedicated center. Medicine (Baltimore). 2003;82:60-76.
42. Aktulga E, Altac M, Müftüoglu A, et al. A double blind study of colchicine in Behçet's disease. Haematologica. 1980;65:399-402.
43. Yurdakul S, Mat C, Tüzün Y, et al. A double-blind trial of colchicine in Behçet's syndrome. Arthritis Rheum. 2001;44:2686-2692.
44. Yazici H, Pazarli H, Barnes CG, et al. A controlled trial of azathioprine in Behçet's syndrome. N Engl J Med. 1990;322:281-285.
45. Hamuryudan V, Ozyazgan Y, Hizli N, et al. Azathioprine in Behçet's syndrome: effects on long-term prognosis. Arthritis Rheum. 1997;40:769-774.
46. Masuda K, Nakajima A, Urayama A, et al. Double-masked trial of cyclosporin versus colchicine and long-term open study of cyclosporin in Behçet's disease. Lancet. 1989;1:1093-1096.
47. Sfikakis PP, Markomichelakis N, Alpsoy E, et al. Anti-TNF therapy in the management of Behçet's disease-review and basis for recommendations. Rheumatology (Oxford). 2007;46:736-741.
48. Melikoglu M, Fresko I, Mat C, et al. Short-term trial of etanercept in Behçet's disease: a double blind, placebo controlled study. J Rheumatol. 2005;32:98-105.
49. Mat C, Yurdakul S, Uysal S, et al. A double-blind trial of depot corticosteroids in Behçet's syndrome. Rheumatology (Oxford). 2006;45:348-352.
50. Hatemi G, Silman A, Bang D, et al. EULAR recommendations for the management of Behçet disease. Ann Rheum Dis. 2008;67:1656-1662.