Article
Risankizumab: A New Treatment Option for Crohn’s Disease
Author(s):
Risankizumab represents the first ever IL-23 treatment for adult patients with Crohn's disease approved by the FDA.


Shubha Bhat, PharmD, MS, BCACP

In June, risankizumab-rzaa (Skyrizi), the first specific interleukin (IL)-23 inhibitor, was approved by the US Food and Drug Administration (FDA) for the treatment of moderate-to-severe Crohn’s disease (CD).
Risankizumab demonstrated significant improvement in clinical and endoscopic responses in two randomized placebo-controlled induction studies, ADVANCE and MOTIVATE, and one maintenance study, FORTIFY.
The ADVANCE clinical trial included 850 patients who were either biologic-naïve (treated with or had intolerance to conventional therapies) or biologic-experienced. On average, participants in this study were 38 years old, male, Caucasian, and had CD for 9 years.


ADVANCE: Risakinzumab (Prior Bio-Failure)
Chart Adapted from Ferrent M, et. Al, The Lancet. 2022. 399(10340):2031-2046.
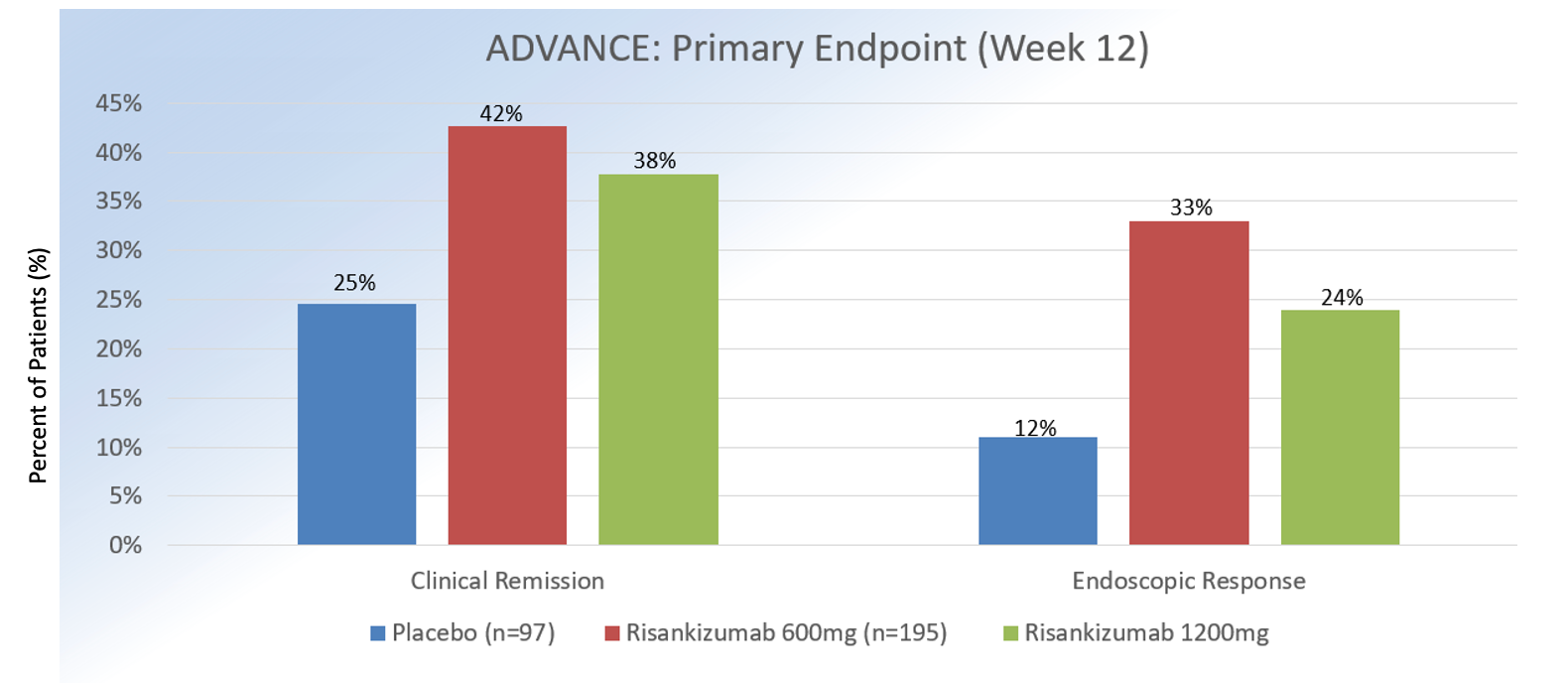
Approximately 58% had been exposed to >1 biologic medication. In MOTIVATE, 569 patients who were biologic-experienced were enrolled. On average, participants were 40 years old, male, Caucasian, and had CD for 9 years.


MOTIVATE: Risakinzumab (Prior Bio-Failure)
Chart Adapted from Ferrent M, et. Al, The Lancet. 2022. 399(10340):2031-2046.
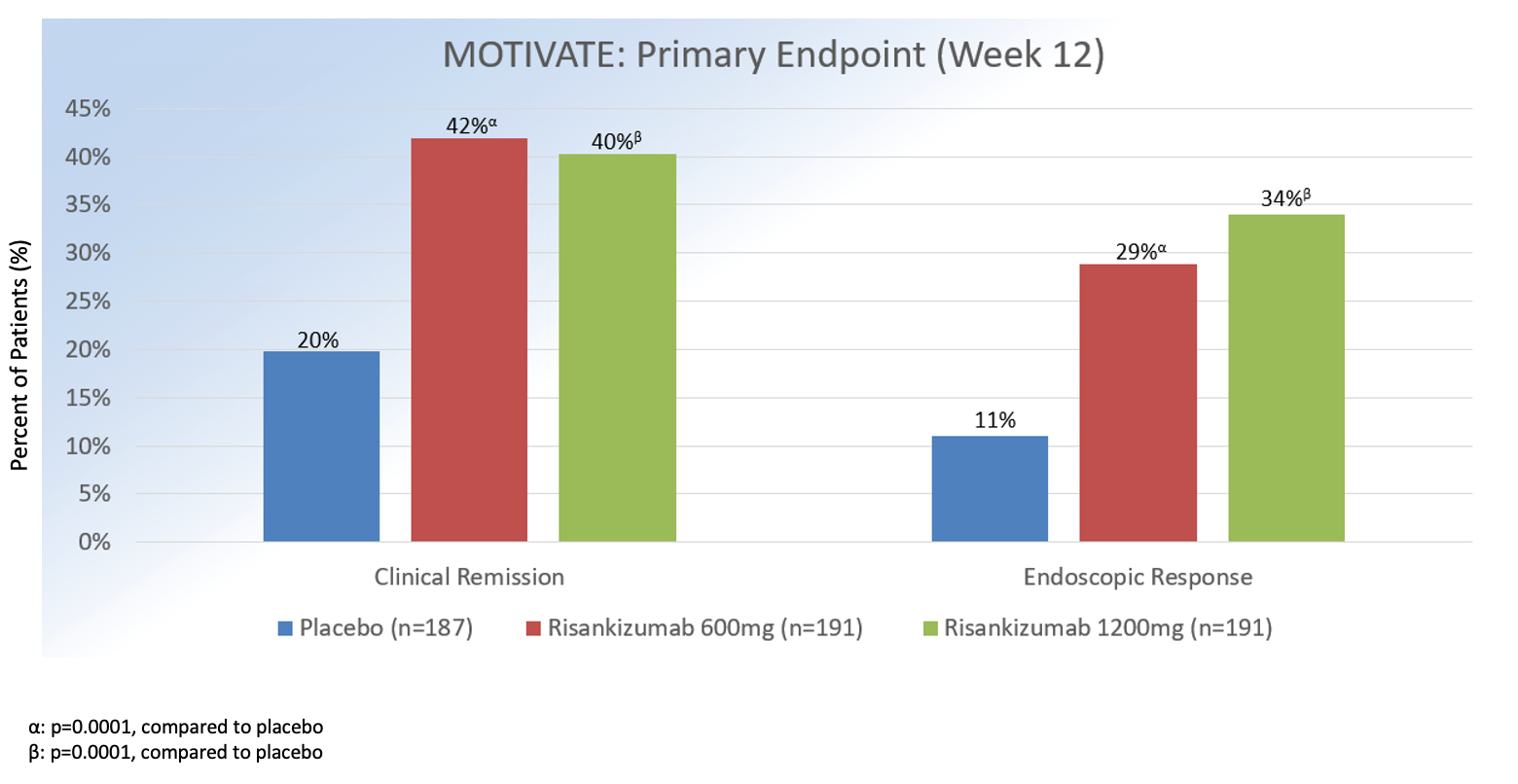
Approximately, 53% had prior exposure to at >2 biologics. In both induction studies, enrollment of patients who had been treated with ustekinumab was capped at 20%.1
At 12 weeks, clinical remission (defined as Crohn’s Disease Activity Index <150 or patient-reported outcomes of average daily liquid or very soft stool frequency of <2.8, plus average daily abdominal pain score of <1) for intravenous (IV) risankizumab 1200 mg versus 600 mg versus placebo was 42%, 45%, and 25% (P = .0001) in ADVANCE and 40%, 42%, and 20% in MOTIVATE (P = .0001).
Endoscopic response (defined as 50% decrease in Simple Endoscopic Score [SES-CD] from baseline or if isolated ileal disease and SES-CD of 4, ≥2 point reduction from baseline) at week 12 for IV risasnkizumab 1200 mg versus 600 mg vs. placebo was 32%, 40%, and 12% in ADVANCE and 34%, 29%, and 11% in MOTIVATE (P = .0001). In regards to safety, the overall incidence of adverse events in ADVANCE and MOTIVATE was similar across all treatment groups, with headaches, nasopharyngitis, and arthralgia being the most commonly reported events.
One patient treated with risankizumab 600 mg in ADVANCE experienced a rash and increase in liver enzymes that resolved with hospitalization and steroid administration.
Although not consistent with drug-induced liver injury, current monitoring parameters for risankizumab involve monitoring liver enzymes and bilirubin levels at baseline and during induction up to 12 weeks of treatment. Overall, risasnkizumab was deemed to be effective and well tolerated as induction treatment. The results of the study are presented in the tables below.
In FORTIFY, 462 patients with clinical response to IV risankizumab from ADVANCE and MOTIVATE were randomized to subcutaneous (SQ) risankizumab 360 mg or 180 mg every 8 weeks or medication discontinuation (placebo). On average, participants were 39 years old, male, Caucasian, and had CD for 9 years.


FORTIFY: Risankizumab for Maintenance
Chart Adapted from Ferrent M, et. Al, The Lancet. 2022. 399(10340):2031-2046.
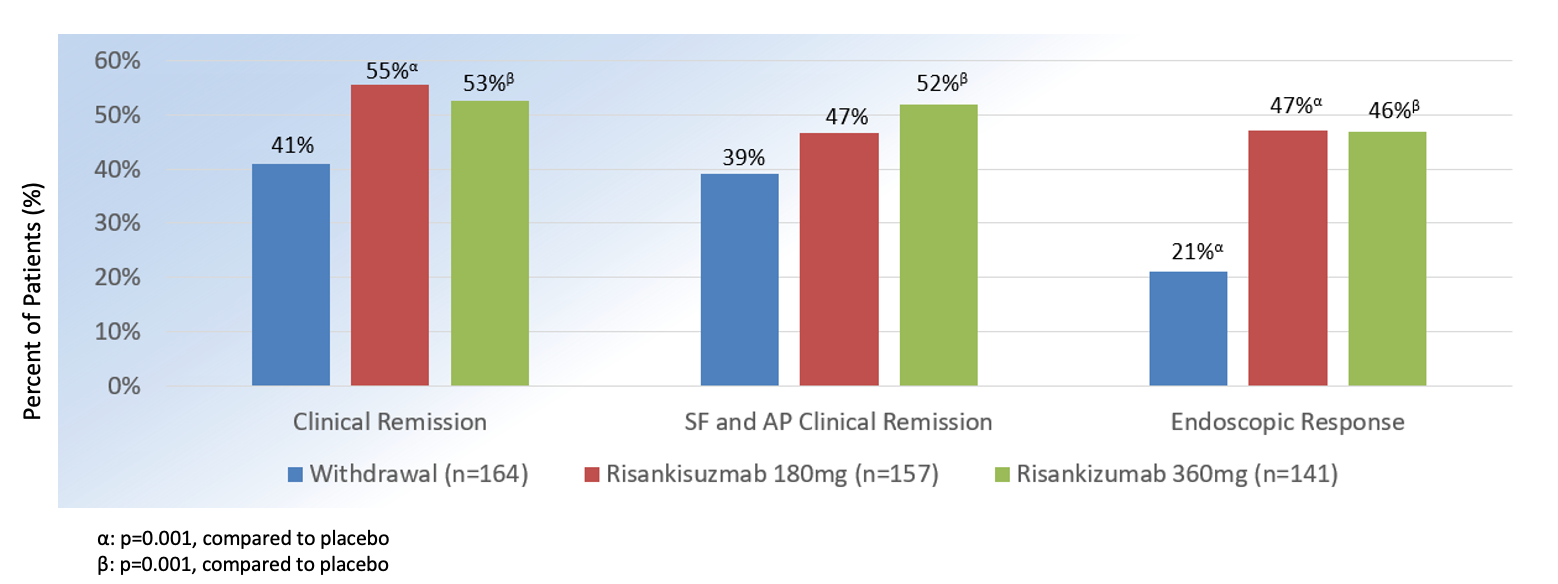
At week 52, rates of clinical remission with risankizumab 360 mg vs. 180 mg vs placebowas 53%, 55%, and 41% (P = .001) and endoscopic response was 46%, 47%, and 21% (P = .001). Risankizumab was overall well tolerated, with most common adverse effects consisting nasopharyngitis, arthralgia, headache, nausea, abdominal pain, diarrhea, anemia, and injection site reactions.2
Of note, the maintenance dose is administered through a novel device known as the on-body injector, which consists of series of steps and requires patients to be trained prior to use to ensure optimal medication delivery.3
With the last therapeutic advance for moderate to severe CD occurring 6 years ago, risankizumab offers a much needed new treatment option for patients. It may also be used for patients with plaque psoriasis and psoriatic arthritis.
Risankizumab’s efficacy rate for CD is comparable to other available treatments, and we await findings from additional studies, such as the ongoing head-to-head study between risankizumab and ustekinumab (an IL-12 and -23 inhibitor), to:
- Understand how to best position use of risankizumab in both biologic-naïve and biologic-experienced patients and
- Further develop our understanding of IBD pathophysiology.
References
- D’Haens G, Panaccione R, Baert F, et al. Risankizumab as induction therapy for Crohn’s disease: results from the phase 3 ADVANCE and MOTIVATE induction trials. The Lancet. 2022;399(10340):2015-2030. doi:10.1016/s0140-6736(22)00467-6
- Ferrante M, Panaccione R, Baert F, et al. Risankizumab as maintenance therapy for moderately to severely active Crohn's disease: results from the multicentre, randomised, double-blind, placebo-controlled, withdrawal phase 3 FORTIFY maintenance trial. Lancet. 2022;399(10340):2031-2046. doi:10.1016/S0140-6736(22)00466-4
- SKYRIZI® (risankizumab-rzaa) Dosing for Crohn’s disease. www.skyrizi.com. Accessed October 10, 2022. https://www.skyrizi.com/crohns/about-skyrizi/dosing#isi

