Article
Allergy and immunology problems and musculoskeletal specialists
Clinicians caring for patients with musculoskeletal disorders may see patients who have underlying conditions that typically are evaluated and managed by allergy and immunology specialists. Although diagnosis of these conditions may be challenging, new insights into their pathogenesis have led to advances in diagnosis and therapy.
Clinicians caring for patients with musculoskeletal disorders may see patients who have underlying conditions that typically are evaluated and managed by allergy and immunology specialists. Although diagnosis of these conditions may be challenging, new insights into their pathogenesis have led to advances in diagnosis and therapy. The ability to recognize allergy and immunology disorders and initiate evaluation and management of patients who have them can improve patient care and, when indicated, facilitate early referral.
In this article, we provide a brief overview of the approach to evaluating patients who have urticaria, angioedema, and adult-onset primary immunodeficiencies when they present to musculoskeletal medicine specialists. We then highlight important concepts about the causes and initial assessment and treatment.
URTICARIA AND ANGIOEDEMA
Urticaria is a common condition that affects more than 15% to 25% of adults at some point in their lifetime.1 It is characterized by transient, well-circumscribed wheals with erythema that are intensely pruritic.


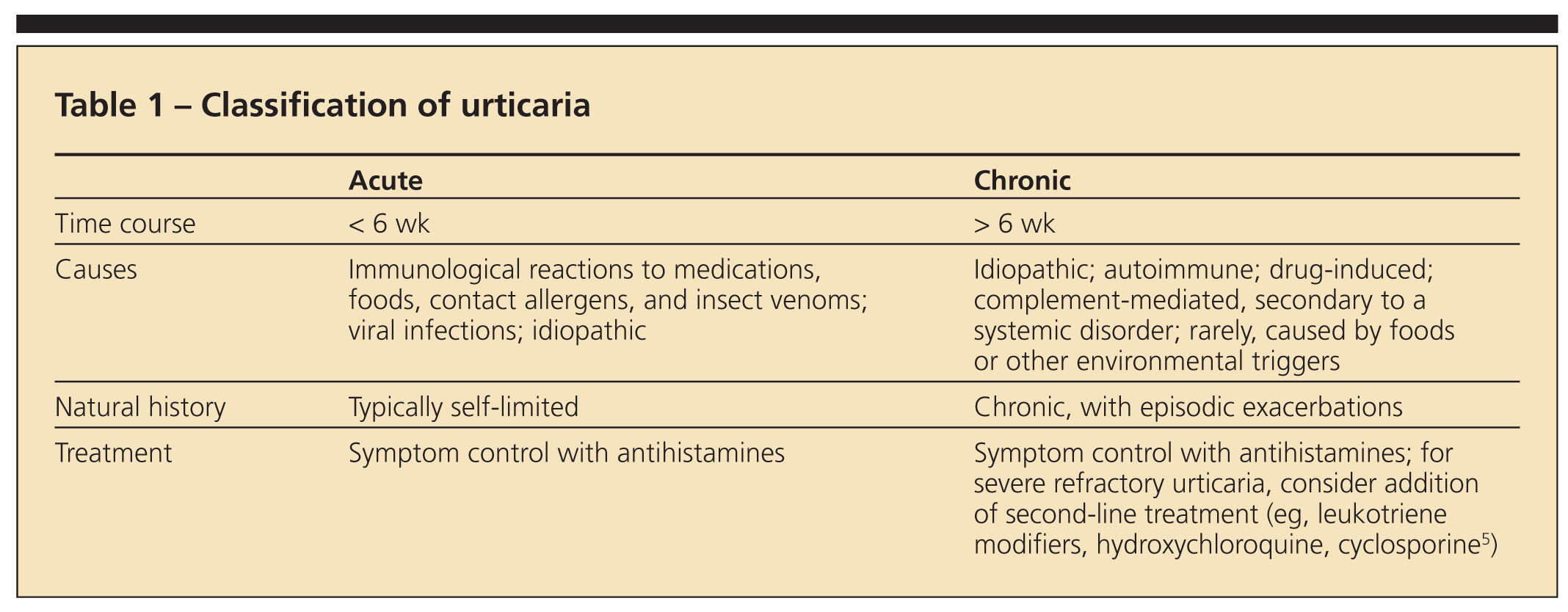
Urticaria may be classified as acute or chronic (Table 1). Most patients with urticaria experience acute urticaria (AU), defined as involving eruptions that last less than 6 weeks. The cause of AU may be an immunological reaction to medications, foods, insect venoms, or contact allergens, but it also can be associated with viral and parasitic infections. Frequently, the inciting trigger is not identified.
Although AU warrants careful evaluation for new exposures or other symptoms that suggest an infection, it rarely necessitates costly laboratory studies. Most cases are self-limited and may be managed with antihistamines alone.
Chronic urticaria
Chronic urticaria (CU) is characterized by more than 6 weeks of symptoms present on most days of the week; it has multiple causes. Immunological reactions to external triggers (eg, foods and medications) are less likely to occur in this group; they are found in fewer than 10% of patients.2 Some drugs (eg, opioid analgesics) can elicit urticaria through direct mast cell activation.
Aspirin and NSAIDs may cause urticaria through various mechanisms, including IgE-mediated hypersensitivity reactions and alterations in arachidonic acid metabolism that shift the balance of prostaglandins and leukotrienes. This shift favors leukotriene generation, a mediator involved with urticaria formation.
Physical forms of urticaria may occur after exposure to water, vibration, temperature changes, mechanical pressure, and solar rays. Diagnosis of these rare forms is made with careful questioning of the patient.


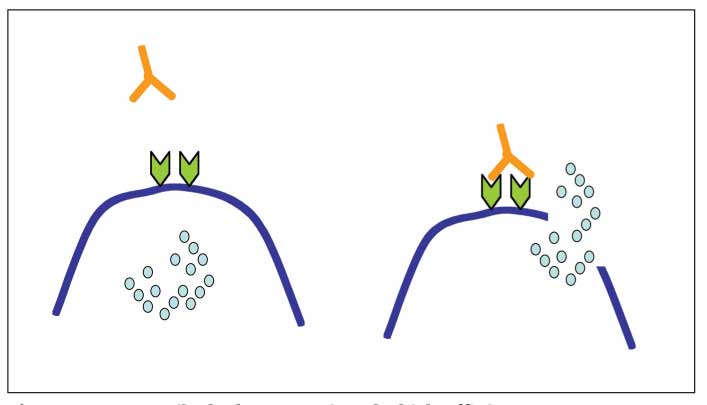
Figure 1 – Autoantibody that recognizes the high-affinity type I IgE Fc receptor can bind and cross-link the receptor on mast cells and basophils, leading to cell activation and release of mediators that cause urticaria or angioedema.
Antigen-antibody complexes and complement activation that occur in connective tissue diseases may activate mast cells, in part through C5a receptors. In addition, more than 40% of patients with chronic idiopathic urticaria (CIU) have autoantibodies directed against the a subunit of the IgE receptor (FceRI) or to cell-surface IgE that may lead to IgE receptor cross-linking (Figure 1).2
Recent studies also have suggested that patients with CIU may have dysregulated T-cell cytokine production that promotes cutaneous inflammation.3 However, the cause of CIU remains unknown in a large proportion of affected patients.
Systemic disorders with urticaria
Various systemic disorders can manifest with CU, including systemic lupus erythematosus (SLE), malignancies, bullous pemphigoid, occult infections, autoinflammatory diseases, and urticarial vasculitis (UV). A thorough history and physical examination are important tools in screening for these underlying disorders; however, no underlying systemic illness is found in most cases of CU.
Several features help differentiate vasculitic lesions from typical urticaria, including persistence of a discrete urticarial lesion beyond 24 hours, a burning or painful sensation instead of pruritus, and associated systemic symptoms (eg, fever, arthralgia, and abdominal pain). A skin biopsy is essential for any lesion suspicious for vasculitis and typically demonstrates leukocytoclastic vasculitis with neutrophilic infiltrates.4
UV is categorized into normocomplementemic and hypocomplementemic subtypes. The latter is more common in patients with systemic rheumatologic disease and those with autoantibodies to C1q.
The history is critical
Critical to identifying potential causative triggers of urticaria is a detailed history that includes recent medication changes, environmental exposures, and associated symptoms. Subsequent laboratory evaluation should be driven by history and physical examination findings.
Although there is no consensus about screening laboratory tests, a reasonable start would include a complete blood cell count with a differential to evaluate for eosinophilia, the thyroid-stimulating hormone test, liver function tests, and erythrocyte sedimentation rate or C-reactive protein level. A serum protein electrophoresis may be considered in older patients who may be at increased risk for monoclonal gammopathies. Musculoskeletal symptoms, photosensitivity, oral ulcers, and serositis symptoms should raise concern for SLE; obtaining a urinalysis, assessing renal function, and measuring antinuclear antibodies is reasonable.
In most cases, screening laboratory test results are normal. In patients with CIU, testing for autoantibodies to the IgE receptor is now available. Evaluation of patients with suspected UV begins with a skin biopsy; if the result is abnormal, a thorough laboratory appraisal for systemic inflammatory disease, occult infection, or autoimmunity is recommended.
Symptom control is the goal
The primary goal of urticaria management is symptom control. Antihistamines are the mainstay of therapy; a combination of sedating and nonsedating antihistamines often is beneficial.
For patients with symptoms refractory to antihistamines alone, second-line treatments include leukotriene modifiers, hydroxychloroquine (HCQ), and cyclosporine (CyA).5 Corticosteroids almost always eliminate urticaria, but they generally should be avoided given their significant adverse effects with prolonged use and the high probability of urticarial flares after discontinuation.
Management of UV is driven by disease severity. Antihistamines are used to control pruritus; corticosteroids, dapsone, HCQ, colchicine, methotrexate, azathioprine, and CyA are other options for disease modification for physicians experienced in their use.


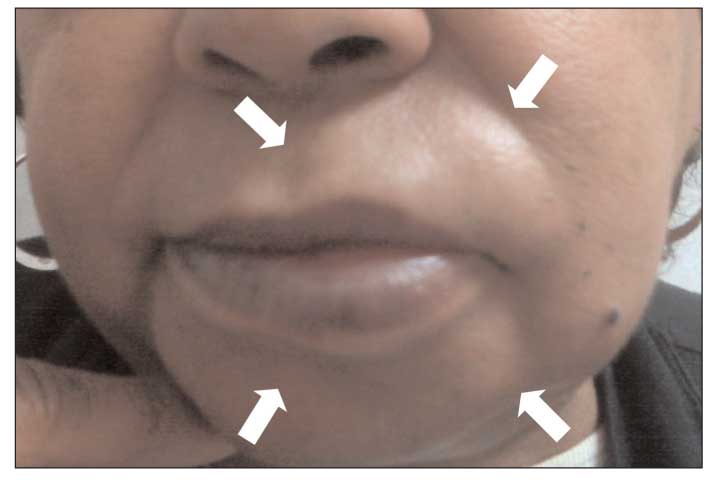
Figure 2 – Angioedema of the lips (arrows) is shown in this photograph.
Deeper skin involvement with angioedema
Unlike urticaria, angioedema is associated with deeper dermal and subcutaneous edema, most often involving the lips (Figure 2), eyelids, larynx, and GI system. Many of the mechanisms that lead to urticaria also cause angioedema; they include acute IgE-mediated hypersensitivity reactions, direct mast cell activation, aspirin- and NSAID-induced alterations in arachidonic acid metabolism, complement-mediated mast cell degranulation, autoantibody-induced mast cell activation, and idiopathic urticaria/angioedema (Table 2).



Patients with UV may present with concurrent angioedema. Note, however, that angioedema also may occur independent of urticaria. Forms of isolated angioedema thought to be mediated by bradykinin include angioedema secondary to angiotensin-converting enzyme (ACE) inhibition, hereditary angioedema (HAE), and acquired angioedema.
In some patients, ACE inhibition may lead to elevated levels of bradykinin with subsequent angioedema because of increased vascular permeability.6 This type of angioedema may occur at any time, even years after patients start taking the drug.
Consider hereditary angioedema
Patients with HAE types I and II have episodic angioedema resulting from abnormal C1 inhibitor levels or function. C1 inhibitor has many functions, including regulating the complement and contact systems to prevent excessive bradykinin generation.7
With HAE type I, the level of C1 inhibitor typically is low, even when the patient is asymptomatic. With HAE type II, the baseline level of C1 inhibitor may be normal or elevated, but the C1 inhibitor function is reduced. Both forms of HAE result in aberrant regulation of bradykinin formation.
The newly described HAE type III is thought to be mediated by alterations in the contact system and appears to be exacerbated by estrogen.8 Acquired angioedema-the condition resulting from production of an inhibitor of C1 inhibitor-typically occurs in the setting of an underlying hematological malignancy.
Evaluation of angioedema
Evaluation begins with a thorough investigation of potential external triggers, including medications (eg, opioid analgesics), acetylsalicylic acid, NSAIDs, and ACE inhibitors), and notation of any associated symptoms. Acute angioedema requires assessment of the patient’s respiratory status to ensure that the patient does not warrant immediate treatment. For patients with recurring angioedema and urticaria, the assessment is the same as for CU.
With HAE types I and II, the C4 level is depressed in most cases, even when the patient is asymptomatic, and serves as a good screening laboratory test. If the baseline C4 level is low, the C1 inhibitor antigenic level and function should be obtained. No associated laboratory abnormalities have been found in patients with HAE type III. In acquired angioedema, C2, C4, and C1q levels typically are low at baseline and C1 inhibitor function is abnormal.
Therapeutic goals
Management of angioedema involves avoidance of identified triggers. For acute IgE-mediated angioedema, antihistamines and corticosteroids are the treatments of choice. Epinephrine may be required for angioedema in the setting of anaphylaxis. Daily suppressive antihistamines may be required in patients with CU and angioedema.
Management of HAE is complicated by a paucity of available effective interventions. However, various novel treatments are under development, including C1 replacement, kallikrein inhibition, and bradykinin receptor antagonists. Long-term prophylaxis for HAE can be achieved with attenuated androgens.9 Acquired angioedema may improve with management of the underlying disease.
HUMORAL IMMUNODEFICIENCY
Patients who report frequent infections often are seen in adult medicine clinics, although the estimated prevalence of primary immunodeficiencies in the adult population is somewhat low. Most clinically important immunodeficiencies in adult patients are the result of other medical conditions, including metabolic disorders, infections, and malignancies, as well as drug effects from immunosuppressive agents. Selective IgA deficiency and common variable immunodeficiency (CVID) are 2 primary immunodeficiencies that may present in adulthood.
IgA deficiency most common
IgA deficiency is the most common primary immunodeficiency; the estimated prevalence is 1 per 500 persons in the United States.10 The vast majority of patients with this condition are asymptomatic. In some patients, however, recurring infections develop with encapsulated bacteria; they present as frequent sinopulmonary infections or unexplained bronchiectasis. Other patients experience anaphylactic reactions during blood product administration because of the presence of endogenous antibodies to residual IgA in transfusions.
Conditions that may be associated with IgA deficiency include several autoimmune diseases (rheumatoid arthritis [RA], SLE, pernicious anemia, and celiac disease) and chronic diarrhea, Giardia infection, and gastric malignancies. Screening laboratory tests include total IgG, IgM, and IgA levels. If a low level or the absence of IgA is confirmed by repeated testing in a patient with recurring infections, referral to an immunologist for further evaluation of immune function is warranted. The diagnosis is confirmed by low levels of IgA (less than 7 mg/dL) in the setting of normal IgG and IgM without other identified causes of immunoglobulin deficiency.
CVID develops in a small percentage of patients with IgA deficiency, although there are no consistent clinical indicators identifying progressors. No treatment is warranted for asymptomatic patients. Antibiotics may be used for infections and chronic suppressive antibiotics for recurring disease. Immunoglobulin replacement is not indicated because it will not replace IgA.
CVID disease features
The prevalence of CVID is estimated at between 1 per 10,000 and 1 per 200,000 persons; disease onset peaks in childhood and early adulthood.11 Common clinical manifestations include recurring sinopulmonary infections, granulomatous disease, chronic diarrhea, autoimmunity, and arthritis, as well as an increased risk of malignancy and laboratory findings of a reduced IgG level with low or absent IgA or IgM. Frequent bacterial infections causing bronchitis, pneumonia, sinusitis, otitis, meningitis, and sepsis are common; they typically involve Streptococcus pneumoniae or Haemophilus influenzae.
Granulomatous lung disease with noncaseating granulomas that resembles sarcoid is evident in 8% to 22% of patients who have CVID.12 Bronchiectasis also may develop as a result of recurring bronchial infections. GI manifestations include symptoms that mimic inflammatory bowel disease, malabsorption, and chronic diarrhea.
More than 20% of patients with CVID have an associated autoimmune condition.12 The most common ones include autoimmune hemolytic anemia, immune thrombocytopenia, and pernicious anemia. CVID also may present with an inflammatory, aseptic, nonerosive polyarthritis typically involving large joints, such as the knees, ankles, and wrists, that can appear similar to RA.13 In addition, patients with CVID are at increased risk for non-Hodgkin B-cell lymphomas and gastric carcinomas. Immunological defects in CVID include impaired B-cell activation after antigen cross-linking, reduced numbers of CD27+ memory B cells, decreased B-cell survival, abnormal T-cell proliferation, and altered T-cell cytokine production culminating in decreased production of immunoglobulins.
Making the diagnosis
The key to diagnosis involves including CVID in the differential diagnosis for patients with unusual or recurrent infections, atypical inflammatory arthritis, or immune cytopenias. Many patients are symptomatic for years before the diagnosis because the condition often goes unrecognized.
The diagnostic evaluation begins with a quantitative analysis of serum immunoglobulin levels, including IgG, IgM, IgA, and IgE. If the levels are low, secondary causes of hypogammaglobulinemia (eg, an occult malignancy, adverse effects of medications, viral infections, and protein-losing conditions) should be excluded. Measurement of isohemagglutinin and specific antibody levels also should be obtained.
Tetanus and pneumococcal polysaccharide antibody levels may be assessed at baseline and 4 to 6 weeks after revaccination. A normal response to the pneumococcal vaccine is a 4-fold increase in the pneumococcal antibody titers in at least 70% of the vaccine serotypes14; an increase in the tetanus toxoid antibody titer also should be observed. A diagnosis of CVID can be made when the serum IgG and IgA (or IgM) levels are less than 2 standard deviations below the mean in the setting of poor antibody responses to vaccinations and exclusion of other causes of hypogammaglobulinemia.
Goals of treatment
Treatment goals for CVID include antibody replacement with intravenous or subcutaneous immunoglobulin, managing infections, monitoring lung function, managing autoimmune disorders, and regular monitoring for malignancy. Noninfectious arthritis may respond well to antibody replacement and treatment with NSAIDs. Persistence of joint pain in the setting of progressive erosive or inflammatory disease warrants evaluation for other possible causes. Management of autoimmune disease may be complicated by the need for immunosuppressive agents, which should be used cautiously with input from a rheumatologist and an immunologist.
References:
References1. Amar SM, Dreskin SC. Urticaria. Prim Care. 2008;35:141-157, vii, viii.
2. Kaplan AP. Chronic urticaria: pathogenesis and treatment. J Allergy Clin Immunol. 2004;114:465-475.
3. Dos Santos JC, Azor MH, Nojima VY, et al. Increased circulating pro-inflammatory cytokines and imbalanced regulatory T-cell cytokines production in chronic idiopathic urticaria. Int Immunopharmacol. 2008;8:1433-1440.
4. Mehregan DR, Hall MJ, Gibson LE. Urticarial vasculitis: a histopathologic and clinical review of 72 cases. J Am Acad Dermatol. 1992;26(3, part 2):441-448.
5. Morgan M, Khan DA. Therapeutic alternatives for chronic urticaria: an evidence-based review, part 1. Ann Allergy Asthma Immunol. 2008;100:403-414, 468.
6. Malde B, Regalado J, Greenberger PA. Investigation of angioedema associated with the use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. Ann Allergy Asthma Immunol. 2007;98:57-63.
7. Davis AE 3rd. Hereditary angioedema: a current state-of-the-art review, III: mechanisms of hereditary angioedema. Ann Allergy Asthma Immunol. 2008;100(1,suppl 2):S7-S12.
8. Cichon S, Martin L, Hennies HC, et al. Increased activity of coagulation factor XII (Hageman factor) causes hereditary angioedema type III. Am J Hum Genet. 2006;79:1098-1104.
9. Zuraw BL. Hereditary angioedema: a current state-of-the-art review, IV: short- and long-term treatment of hereditary angioedema: out with the old and in with the new? Ann Allergy Asthma Immunol. 2008;100(1, suppl 2):S13-S18.
10. Rosen FS, Cooper MD, Wedgwood RJ. The primary immunodeficiencies (1). N Engl J Med. 1984;311:235-242.
11. Cunningham-Rundles C. Common variable immunodeficiency. Curr Allergy Asthma Rep. 2001;1:421-429.
12. Di Renzo M, Pasqui AL, Auteri A. Common variable immunodeficiency: a review. Clin Exp Med. 2004;3:211-217.
13. Swierkot J, Lewandowicz-Uszynska A, Chlebicki A, et al. Rheumatoid arthritis in a patient with common variable immunodeficiency: difficulty in diagnosis and therapy. Clin Rheumatol. 2006;25:92-94.
14. Bonilla FA, Bernstein IL, Khan DA, et al. Practice parameter for the diagnosis and management of primary immunodeficiency [published correction appears in Ann Allergy Asthma Immunol. 2006;96:504]. Ann Allergy Asthma Immunol. 2005;94(5, suppl 1):S1-S63.
















