Article
Identifying and managing hemochromatosis arthropathy
Hemochromatosis is a disorder of iron metabolism that may lead to abnormalities in multiple organ systems.
Hemochromatosis is a disorder of iron metabolism that may lead to abnormalities in multiple organ systems. It is characterized by excessive body iron stores and deposition of hemosiderin, which can cause tissue damage and organ dysfunction. The diagnosis is often delayed, sometimes for decades, because a period of silent iron deposition occurs before symptoms are manifest. Diagnosis is difficult even in symptomatic patients, because the signs and symptoms are nonspecific and overlap with those of many other conditions.
Joint abnormalities are common and often lead to morbidity and loss of quality of life. Joint disease often is an early manifestation of hemochromatosis and may be the symptom complex that leads to hemochromatosis diagnosis; however, a high index of suspicion must be maintained because the joint findings mimic those seen in other common rheumatologic conditions.
In this article, we briefly describe the diagnosis and management of hemochromatosis. Then we focus on the findings in hemochromatosis arthropathy and those of several conditions with similar symptoms involved in the differential diagnosis, including rheumatoid arthritis (RA), osteoarthritis (OA), and calcium pyrophosphate dihydrate (CPPD) crystal deposition disease.
HEMOCHROMATOSISGenetics and classification
After elucidation of the genetics of hemochromatosis, the disorder is now best defined as the presence of 2 mutations of the hemochromatosis (HFE) gene.1 Subsequent development of iron overload, which is variable among persons with the mutations, can cause complications in multiple organ systems.
Several mutations of the HFE gene have been defined. In the United States, homozygosity for the C282Y mutation accounts for most clinical cases of iron overload2,3 This mutation may have arisen from a single progenitor in northwestern Europe.2 H63D is the next most common mutation, followed by S65C. Compound heterozygosity of C282Y and H63D accounts for a very few additional cases.1,2 The C282Y mutation results in abnormal binding of the HFE protein to β2-microglobulin, leading to excessive iron absorption.
Hemochromatosis type 1 is the common adult type of hemochromatosis (mutations of HFE). Hemochromatosis type 2 (juvenile hemochromatosis) is divided into type 2A (mutations of the hemojuvelin gene, HJV) and type 2B (mutations of the hepcidin antimicrobial peptide gene, HAMP). Hemochromatosis types 3 through 5 are associated with mutations that inactivate transferrin receptor 2 (TfR2), mutations of the ferroportin gene (SLC11A3), and mutations of the H ferritin gene, respectively. Intensive investigation is under way to determine the role of each of these genes and their mutations and also of the iron-regulated gene 1, the duodenal cytochrome b (DCYTB, CYBYRD1), and others in iron accumulation.
Symptoms and clinical findings
Symptoms of hemochromatosis result from damage to various organs caused by iron overloading. Symptoms tend to appear earlier in affected men than in women.2,4 Patients whose hemochromatosis is diagnosed on the basis of symptoms have evidence of iron overload and end-organ damage; those with hemochromatosis diagnosed by screening manifest fewer symptoms, if any (Table 1).4,5 Because of a trend of earlier diagnosis, the classic symptoms of cirrhosis, diabetes mellitus (DM), and bronze skin are seen infrequently; the most common presenting symptoms are fatigue, malaise, and joint symptoms.2
Many organs may be affected by iron overload. Eventually, overload in the liver may lead to cirrhosis and hepatocellular carcinoma. Cardiac iron deposition may lead to cardiomyopathy, with arrhythmia and congestive heart failure. Deposition of iron in the pituitary gland can lead to hypogonadrotropic hypogonadism, hypothyroidism or, rarely, panhypopituitarism. Deposition of iron in the islet cells of the pancreas may lead to DM. The skin may take on a bronze hue (Figure 1). Joint manifestations are common.


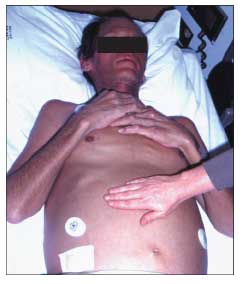
Figure 1 – The skin of this patient with hemochromatosis exhibits the bronze pigmentation characteristic of the disease (compare with the skin of the unaffected person's hand in the foreground). The patient also demonstrates abdominal distention and an everted umbilicus resulting from tense ascites; this complicates cirrhosis caused by hepatic iron deposition.
Laboratory findings
The most common laboratory abnormalities in hemochromatosis are elevations of serum iron concentration, percent saturation of transferrin, and serum ferritin concentration.4 Often, transferrin saturation is the first laboratory abnormality observed; it may be detected in some children and teenagers with the disease.2,4,6 Values for ferritin concentration and transferrin saturation in patients with clinical hemochromatosis compared with asymptomatic persons who were identified by screening are shown in Table 2.4 A transferrin saturation of 60% or more in symptomatic men or 50% or more in symptomatic women is highly sensitive for the diagnosis.
There is debate about the advisability of screening the general population for hemochromatosis. In 2006, the US Preventive Service Task Force recommended against routine genetic screening for hemochromatosis in asymptomatic persons.7 An NIH-sponsored screening study was performed and reported recently.8 In this study (Hemochromatosis and Iron Overload Screening [HEIRS]), 100,000 persons in North America were screened for hemochromatosis in a primary care setting. Very few newly diagnosed C282Y homozygotes had any symptoms of illness.
It seems prudent to evaluate the siblings of hemochromatosis homozygotes for the presence of 2 mutations of HFE-C282Y and H63D. Siblings who are homozygous for the C282Y mutation should be evaluated for iron overload, employing transferrin saturation and serum ferritin concentration. Siblings who are not homozygous for the C282Y mutation do not need to undergo further evaluation or follow-up for hemochromatosis. Compound heterozygotes usually do not become iron-loaded.
Other common laboratory findings reflect sequelae of damage to organs from iron overload. These include elevated alanine aminotransferase and aspartate aminotransferase levels (usually to 2 to 5 times the upper limit of normal), abnormalities of serum glucose or thyroid-stimulating hormone, and hypogonadotropic hypogonadism. An ECG may show arrhythmia, and decreased ejection fraction may be present on an echocardiogram.
Liver biopsy is very informative in selected patients, but many persons do not need to undergo the procedure. In young homozygotes whose serum ferritin level is normal, liver histology is expected to be unaffected by iron overload and should not show fibrosis or cirrhosis in the absence of another liver disease. Liver biopsy is not needed in such persons, because it will not provide important information or change therapy and it poses risk and expense. Cirrhosis is not likely to be present in patients whose serum ferritin level is lower than 1000 ng/mL or whose hepatic iron concentration is below 22,000 μg/g of liver wet weight.1,9 In patients with high levels of serum ferritin, liver biopsy with quantitation of hepatic iron concentration can be helpful in planning iron depletion therapy. Patients who have massive hepatic iron overload may be served best by phlebotomy of 2 units of whole blood each week until iron depletion is accomplished.
Treatment
Phlebotomy is the mainstay of treatment, both for initial depletion of excess iron stores and for subsequent maintenance of normal total body iron. During initial iron depletion therapy, scheduled phlebotomy generally consists of removal of 500 mL of whole blood weekly until the hematocrit falls 2 to 4 mL/dL below baseline. Smaller volumes (eg, 250 mL) may be removed in fragile patients. Twice-weekly phlebotomy may be considered in those who have very severe iron overload noted on liver biopsy (Figure 2) or in patients who have liver failure or heart failure.


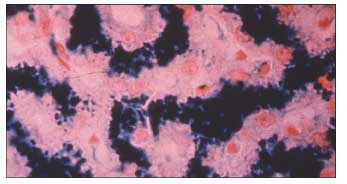
Figure 2 – The liver biopsy specimen seen here shows severe (grade 4) hepatic iron deposition.
Initial iron depletion is followed by lifelong phlebotomy to maintain normal total-body iron. Maintenance phlebotomy involves removal of 500 mL of whole blood every 2 to 4 months in men or every 3 to 12 months in women before menopause. The intent of lifelong maintenance phlebotomy therapy is to allow persons to maintain a normal hematocrit level with a serum ferritin concentration of about 50 ng/mL. A ferritin concentration that rises to greater than 100 ng/mL suggests inadequate iron removal and the need for more frequent phlebotomy therapy.
Patients with hemochromatosis should avoid ingestion of hepatotoxins, such as alcohol. Alcohol can increase iron absorption, and some red wines have a high iron content.10,11 There are some data indicating that ingestion of more than 30 g of ethanol per day may worsen the hepatic injury that is caused by iron overload 12,13 and may increase the risk of hepatocellular carcinoma in patients who have cirrhosis.12
Patients with hemochromatosis should not eat raw oysters because of the risk of septicemia and death from Vibrio vulnificus infection.14-17 Iron-rich foods need not be restricted from the diet, because phlebotomy therapy is a much more efficient process than dietary limitations. Restriction of vitamin C is suggested by some authors, advice most applicable to patients with hemochromatosis cardiomyopathy, in whom vitamin C ingestion can increase iron absorption as well as the tendency toward ventricular arrhythmia.18,19 Medicinal iron supplementation should be discouraged.
Some patients experience improvement in joint symptoms after iron depletion therapy, but the majority do not. In some patients, the first onset of arthralgia occurs after phlebotomy therapy is initiated.20-22 Currently, predicting which persons will experience improvement or worsening of joint pain after iron depletion is not possible.
NSAIDs and acetaminophen are useful for relief of arthralgia in hemochromatosis, but they should not be used in patients who have cirrhosis or renal failure. Older persons may be more sensitive to the hepatotoxic and nephrotoxic effects of nonsteroidal medications than younger patients.
RHEUMATOLOGIC COMPLICATIONS
An arthropathy associated with hemochromatosis was first described by Schumacher in 1964.23 The rate of occurrence of arthritis in persons with hemochromatosis has been variously estimated depending on the design of the study. Rates of about 50% have been reported when rigorous criteria are used to define arthritis,24 and rates have varied in other survey-based studies.25-28
A survey of C282Y homozygotes compared with controls in whom the same survey instrument was administered (to reduce ascertainment bias) showed no statistically significant difference in the prevalence of joint complaints-82% of C282Y homozygotes answered "yes" to the question, "Do you have chronic pain in any of your joints?" compared with 62% of the controls. Homozygotes were more likely than controls to report symptoms in the fingers or hands (50% vs 34%), hips (31% vs 21%), and knees (71% vs 52%). Although none of these differences reached statistical significance, the small sample size of the homozygous population limited the study's power.29
In the HEIRS study, the complaint of arthralgia was not more common among C282Y homozygotes compared with normal study participants.8 Because the symptoms and signs of hemochromatosis arthritis can overlap with those of other common joint disorders, the diagnosis can be overlooked.1,6,22,24,30,31
Pathogenesis
Iron overload is postulated to be the cause of joint damage in patients who have hemochromatosis arthropathy, but the mechanism remains unclear. Iron or ferritin may be found in synovial type A and type B cells and macrophages within joints, but this also may be true in other types of arthritis.22,31
Apatite and CPPD crystals are found in some patients who have hemochromatosis arthropathy. In fact, the pattern of arthritis often can imitate pseudogout.31 In animal models, intra-articular iron has been shown to reduce CPPD crystal clearance from joints.32 However, hemochromatosis arthropathy often is observed without associated crystals. When found, crystals are not morphologically associated with iron or ferritin deposits.22


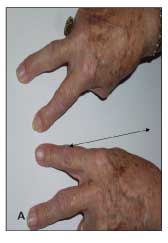
Figure 3 – Note the prominent enlargement of the second and third metacarpophalangeal joints (arrows) in the fingers of this patient with hemochromatosis arthritis who is making the "victory sign" while attempting to fully approximate the second and third fingers (A). The loss of depressions between knuckle ridges of these joints (arrows) is notable when the same patient's closed fists are viewed (B).


Symptoms and signs
Most patients with symptomatic arthropathy of hemochromatosis present with chronic, indolent pain and joint stiffness; bony enlargement; and minimal signs of inflammation.22,24,31 Hemochromatosis may involve the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints, wrists, knees, hips, feet, and shoulders. The distribution of affected joints combined with the character of the arthritis offers important clues to the diagnosis. The arthropathy is generally symmetrical and polyarticular. A predilection for disease in the second and third MCP joints is notable, and enlargement of these joints is readily seen in the fingers when they are extended to form a V, as in the "victory" sign (Figure 3).31
Acute episodes of inflammatory arthritis also may occur, and they may be associated with recovery of CPPD crystals from joint fluid aspiration. A rare syndrome of septicemia accompanied by monoarticular or oligoarticular septic arthritis caused by Yersinia species has been described in patients who have hemochromatosis.22 Prosthetic joints seem especially susceptible to this infection.1
Results from case reports have suggested that joint destruction related to repetitive motion or exercise may be particularly prominent in hemochromatosis arthropathy.33,34 It also has been suggested that the diagnosis of hemochromatosis should be entertained when younger patients present with symptoms suggestive of early, severe OA, such as the need for arthroplasty in the fifth decade of life or earlier.6,33
Radiographic findings
The radiographic findings in hemochromatosis are those of a degenerative arthritis with the joint distribution noted above. Early lesions consist of subchondral cysts with sclerotic margins; they may be especially notable at the metacarpal heads. Generalized osteoporosis appears to be common in patients with hemochromatosis.22 Joint-space narrowing occurs as arthritis progresses and generally is asymmetrical with cartilage loss most pronounced near subchondral cysts.31
Subchondral cysts and sclerosis are less common in the knees than in other affected joints. In the hips, cartilage narrowing and marginal osteophytes are common and may be indistinguishable from the radiographic features of OA.24,31


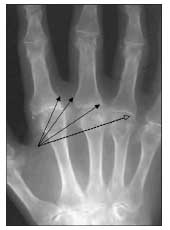
Figure 4 – In this x-ray film, prominent osteophytes can be seen in the second through fifth metacarpophalangeal (MCP) joints (solid arrows). Note the hook shape of the osteophyte in the proximal fifth MCP joint (dashed arrow). Also seen are a marked loss of joint space and partial subluxation of the MCP joints.
Osteophyte formation is common. The hook or beak shape of osteophytes in the MCP joints may help distinguish hemochromatosis from other conditions (Figure 4).22,24,31
Chondrocalcinosis affecting the triangular fibrocartilage above the ulnar styloid, the knees (Figure 5), or the MCP joints is common. Occasionally, chondrocalcinosis in the shoulders, elbows, and other joints may be seen.22,24,31 Not all joints affected by hemochromatosis exhibit chondrocalcinosis, and many joints affected by hemochromatosis arthritis do not have CPPD crystals detected in synovial fluid.31 Profound joint destruction may be visualized in radiographs of the knees and hips. Hemochromatosis arthropathy can result in the need for joint replacement.35


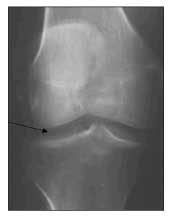
Figure 5 – This x-ray film of the right knee demonstrates chondrocalcinosis (arrow).
Differential diagnosis
There are both clinical and radiographic clues to differentiating between hemochromatosis arthropathy and common overlapping rheumatologic disorders (Table 3).35 In general, the radiographic appearance of individual joints in hemochromatosis arthropathy is similar to that seen in OA or pseudogout, but the distribution of affected joints is reminiscent of other forms of inflammatory arthritis.
Rheumatoid arthritis. The predilection for the MCP, PIP, and wrist joints may suggest RA.36 However, hemochromatosis arthropathy usually is associated with bony enlargement and absence of examination findings of inflammation; RA often presents with inflammatory effusions.
Hemochromatosis arthropathy is not associated with the ulnar deviation of advanced RA.31,36 There have been occasional reports of associated bursitis, tendinitis, and even of subcutaneous nodules.36 Radiographically, the findings of marginal erosions and synovial swelling commonly seen in RA are not seen in hemochromatosis arthritis. Occasionally, a positive rheumatoid factor is reported in patients who have hemochromatosis arthritis.31,36
Because of the overlap of joint distribution, patients thought to have seronegative RA may be proved with iron and DNA testing to have hemochromatosis arthritis. RA and hemochromatosis can coexist, complicating the diagnostic picture. The H63D mutation is more prevalent in patients with RA who have certain HLA epitopes compared with normal subjects, and H63D is a risk factor for the development of RA.1,37,38
Osteoarthritis. The radiographic and physical findings in a given joint affected by hemochromatosis arthritis may be similar to those in OA, but the distribution of joints affected assists in distinguishing the diseases. Like OA, hemochromatosis arthritis commonly affects the PIP joints; however, hemochromatosis arthritis affects the MCP joints while sparing the DIP joints-a pattern inverse to that found in OA.22
Radiographically, the presence of chondrocalcinosis may suggest hemochromatosis arthritis. In the hips, where chondrocalcinosis is unusual and both diseases are common, distinguishing between the 2 may be extremely challenging.31 The results of iron studies and genetic tests, as well as the younger age at onset of arthritis in patients with hemochromatosis, also may help differentiate these diseases.1,22
CPPD disease. Differentiating between idiopathic CPPD disease and hemochromatosis arthritis is perhaps the most challenging. The diseases share many features, including chondrocalcinosis, radiographic appearance and, to a large degree, the distribution of affected joints. About 30% of patients with hemochromatosis arthritis have CPPD crystals recovered from synovial fluid.22,31 Patients with hemochromatosis arthritis also may present with acute flares of inflammatory arthritis.31
Radiographically, CPPD in the wrist is more likely to cause scapholunate dissociation (a loss of the normal anatomical articulation of the ulnar aspect of the scaphoid with the radial aspect of the lunate, manifesting as a malalignment of these bones on an anteroposterior projection). Although hemochromatosis arthritis often affects the second and third MCP joints, it has been reported to affect the fourth and fifth MCP joints more frequently than does idiopathic CPPD disease.22
A hook or beak shape in osteophytes on the radial aspect of the carpal heads also suggests hemochromatosis.1,22,24,31,36 Because there is a substantial overlap of symptoms, it is advisable to test for hemochromatosis when idiopathic CPPD disease is diagnosed.1,31
Treatment
Arthropathy often does not improve after iron depletion therapy, and persistence of joint symptoms is often detrimental to patients' quality of life.1,22 Treatment of patients with arthropathy focuses on control of symptoms and generally is similar to that for those with OA, using analgesics or anti-inflammatory agents, and the effectiveness of several preparations has been studied (Table 4).1,39 However, complications of hemochromatosis in other organ systems, including cardiomyopathy, liver dysfunction, and DM, may influence the choice of agents.
Acute episodes of arthritis may signal acute inflammation with deposition of CPPD crystals. Treatment is the same as that used for acute pseudogout (a potent NSAID, colchicine, or an intra-articular corticosteroid). Joint fluid aspiration should be performed to look for the presence of CPPD crystals and exclude infection. Some patients who experience CPPD crystal deposition may benefit from long-term, low-dose colchicine (0.5 or 1 mg/d). Intra-articular infusion of hyaluronic acid may be somewhat more effective in joints with crystals.22
References:
References1. Edwards CQ. Hemochromatosis. In: Greer JP, Foerster J, Rodgers GM, et al, eds. Wintrobe's Clinical Hematology. 12th ed. Philadelphia: Wolters Kluwer/Lippincott Williams & Wilkins Health. In press.
2. Pietrangelo A. Hereditary hemochromatosis-a new look at an old disease. N Engl J Med. 2004;350:2383-2397.
3. Beutler E, Felitti VJ, Koziol JA, et al. Penetrance of 845G â A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet. 2002;359:211-218.
4. Edwards CQ, Griffen L, Bulaj Z, et al. Estimate of the frequency of morbid complications of hemochromatosis. In: Barton JC, Edwards CQ, eds. Hemochromatosis: Genetics, Pathophysiology, Diagnosis and Treatment. Cambridge, UK: Cambridge University Press; 2000:314.
5. Bulaj ZJ, Ajioka RS, Phillips JD, et al. Disease-related conditions in relatives of patients with hemochromatosis. N Engl J Med. 2000;343:1529-1535.
6. Vaiopoulos G, Papanikolaou G, Politou M, et al. Arthropathy in juvenile hemochromatosis. Arthritis Rheum. 2003;48:227-230.
7. US Preventive Services Task Force. Screening for hemochromatosis: recommendation statement. Ann Intern Med. 2006;145:204-208.
8. Adams PC, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352:1769-1778.
9. Morrison ED, Brandhagen DJ, Phatak PD, et al. Serum ferritin level predicts advanced hepatic fibrosis among US patients with phenotypic hemochromatosis. Ann Intern Med. 2003;138:627-633.
10. Celada A, Herreros V, Pugin P, Rudolf H. Reduced leucocyte alkaline phosphatase activity and decreased NBT reduction test in induced iron deficiency anaemia in rabbits. Br J Haematol. 1979;43:457-463.
11. Conrad ME, Barton JC. Anemia and iron kinetics in alcoholism. Semin Hematol. 1980;17:149-163.
12. Deugnier YM, Guyader D, Crantock L, et al. Primary liver cancer in genetic hemochromatosis: a clinical, pathological, and pathogenetic study of 54 cases. Gastroenterology. 1993;104:228-234.
13. LeSage GD, Baldus WP, Fairbanks VF, et al. Hemochromatosis: genetic or alcohol-induced? Gastroenterology. 1983;84:1471-1477.
14. Bullen J. Bacterial infections in hemochromatosis. In: Barton JC, Edwards CQ, eds. Hemochromatosis: Genetics, Pathophysiology, Diagnosis and Treatment. Cambridge, UK: Cambridge University Press; 2000:381-386.
15. Bullen JJ, Spalding PB, Ward CG, Gutteridge JM. Hemochromatosis, iron and septicemia caused by Vibrio vulnificus. Arch Intern Med. 1991;151:1606-1609.
16. Gerhard GS, Levin KA, Price Goldstein J, et al. Vibrio vulnificus septicemia in a patient with the hemochromatosis HFE C282Y mutation. Arch Pathol Lab Med. 2001;125:1107-1109.
17. Kumamoto KS, Vukich DJ. Clinical infections of Vibrio vulnificus: a case report and review of the literature. J Emerg Med. 1998;16:61-66.
18. McLaran CJ, Bett JH, Nye JA, Halliday JW. Congestive cardiomyopathy and haemochromatosis-rapid progression possibly accelerated by excessive ingestion of ascorbic acid. Aust N Z J Med. 1982;12:187-188.
19. Nienhuis AW. Vitamin C and iron. N Engl J Med. 1981;304:170-171.
20. Edwards CQ, Cartwright GE, Skolnick MH, Amos DB. Homozygosity for hemochromatosis: clinical manifestations. Ann Intern Med. 1980;93:519-525.
21. Adams PC, Speechley M. The effect of arthritis on the quality of life in hereditary hemochromatosis. J Rheumatol. 1996;23:707-710.
22. Faraawi R, Harth M, Kertesz A, Bell D. Arthritis in hemochromatosis. J Rheumatol. 1993;20:448-452.
23. Schumacher HR Jr. Hemochromatosis and arthritis. Arthritis Rheum. 1964,7:41-50.
24. McDonnell SM, Preston BL, Jewell SA, et al. A survey of 2851 patients with hemochromatosis: symptoms and response to treatment. Am J Med. 1999;106:619-624.
25. Ross JM, Kowalchuk RM, Shaulinsky J, et al. Association of heterozygous hemochromatosis C282Y gene mutation with hand osteoarthritis. J Rheumatol. 2003;30:121-125.
26. Timms AE, Sathananthan R, Bradbury L, et al. Genetic testing for haemochromatosis in patients with chondrocalcinosis. Ann Rheum Dis. 2002;61:745-747.
27. Willis G, Scott DG, Jennings BA, et al. HFE mutations in an inflammatory arthritis population. Rheumatology. 2002;41:176-179.
28. Rovetta G, Grignolo MC, Buffrini L, Monteforte P. Prevalence of C282Y mutation in patients with rheumatoid arthritis and spondylarthritis. Int J Tissue React. 2002;24:105-109.
29. Waalen JF, Felitti VF, Gelbart TF, et al. Prevalence of hemochromatosis-related symptoms among individuals with mutations in the HFE gene. Mayo Clin Proc. 2002;77:522-530.
30. Rull M, Schumacher R. The arthropathy of hemochromatosis. In: Barton JC, Edwards CQ, eds. Hemochromatosis: Genetics, Pathophysiology, Diagnosis and Treatment. Cambridge, UK: Cambridge University Press; 2000:258-267.
31. Lambert RE. Iron storage diseases. In: Ruddy S, Harris ED, Sledge CB, eds. Kelley's Textbook of Rheumatology. 6th ed. Philadelphia: WB Saunders; 2001:1559-1565.
32. Brighton CT, Bigley EC Jr, Smolenski BI. Iron-induced arthritis in immature rabbits. Arthritis Rheum. 1970;13:849-857.
33. McCurdie I, Perry JD. Haemochromatosis and exercise related joint pains. BMJ. 1999;318:449-451.
34. Morgan C, Smith D. Haemochromatosis arthropathy and repetitive trauma. Ann Rheum Dis. 2002;61:763.
35. Waalen J, Felitti V, Gelbart T, et al. Prevalence of hemochromatosis-related symptoms among individuals with mutations in the HFE gene. Mayo Clin Proc. 2002;77:522-530.
36. Lonardo A, Neri P, Mascia MT, Pietrangelo A. Hereditary hemochromatosis masquerading as rheumatoid arthritis. Ann Ital Med Int. 2001;16:46-49.
37. Willis G, Scott DG, Jennings BA, et al. HFE mutations in an inflammatory arthritis population. Rheumatology. 2002;41:176-179.
38. Li J, Zhu Y, Singal DP. HFE gene mutations in patients with rheumatoid arthritis. J Rheumatol. 2000;27:2074-2077.
39. Schumacher HR, Straka PC, Krikker MA, Dudley AT. The arthropathy of hemochromatosis. Ann N Y Acad Sci. 1988;526:224-233.
















