Article
Meeting the Challenge of the Vasculitides
The vasculitides are a heterogeneous group of disorders characterized by inflammation and necrosis of blood vessel walls resulting in impaired blood flow and organ damage. This article addresses some of the most frequently asked questions about these conditions.
The vasculitides are a heterogeneous group of disorders characterized by inflammation and necrosis of blood vessel walls resulting in impaired blood flow and organ damage. They have a variety of clinical presentations and several disease mimickers, often have incompletely understood pathophysiology, and are somewhat rare, presenting clinicians with a significant diagnostic challenge. In this article, we attempt to answer some of the most frequently asked questions about these conditions.
How are the vasculitides classified?
The most practical way to classify the vasculitides is according to the size of the predominant vessels that are affected (Table 1). Large-vessel vasculitides involve the aorta and its branches; medium-vessel vasculitides affect medium-size and small arteries of the kidneys, liver, heart, brain, muscles, and GI tract; and small-vessel vasculitides predominantly target capillaries and postcapillary venules.


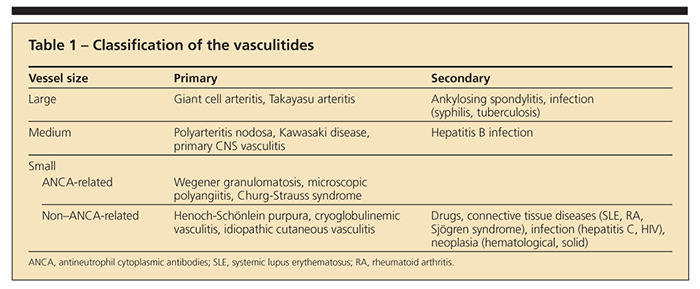
Vasculitides may be primary or occur secondary to drug treatment, connective tissue diseases, infections, or malignancies. They may involve multiple organs (systemic) or be limited to a single organ. Idiopathic and drug-induced cutaneous vasculitis, giant cell arteritis (GCA), and Wegener granulomatosis (WG) are the vasculitides seen most frequently in North America.
When should I suspect a vasculitis?
Many patients with a primary vasculitis present with constitutional symptoms, including fever, night sweats, fatigue, anorexia, and weight loss. Also common are arthralgia, myalgia and, less frequently, arthritis. In the absence of more specific symptoms, the diagnosis often is delayed.


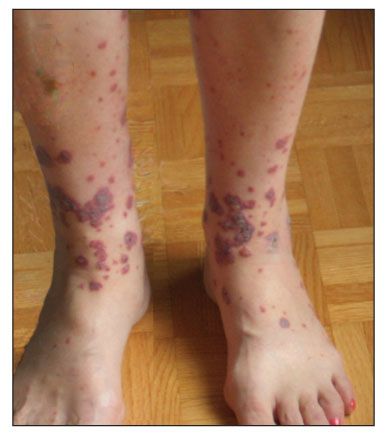
Figure –Idiopathic small-vessel vasculitis with lesions of palpable purpura and hemorrhagic bullae is seen in this photograph.
Palpable purpura is a classic manifestation of the small-vessel vasculitides. Other, less specific cutaneous manifestations include nonpalpable purpura, vesicles, bullae (often hemorrhagic as a result of capillary wall rupture), urticaria (the lesions often are painful rather than pruritic and tend to last longer than 24 hours), superficial erosions, and splinter hemorrhages (Figure).1 More characteristic of involvement of vessels from the deep dermis are livedo reticularis, nodules, and ulcers, which are common in polyarteritis nodosa (PAN).
Another presentation that should raise suspicion of the presence of vasculitis, particularly in patients who do not have diabetes mellitus, is mononeuritis multiplex. Defined as the sudden occurrence of a sensory or motor neuropathy or both in random peripheral nerves (eg, right median and left common peroneal nerve palsies), this disorder results from necrotizing inflammation of nerve blood vessels. Mononeuritis multiplex typically is seen in PAN, as well as in a number of small-vessel vasculitides, including Churg-Strauss syndrome (CSS), cryoglobulinemic vasculitis (CV), WG, microscopic polyangiitis (MPA), and vasculitis complicating rheumatoid arthritis.
The new onset of headache, jaw claudication, or scalp tenderness in an older patient is highly suggestive of GCA, particularly if it is associated with vision symptoms (diplopia, amaurosis fugax) or symptoms of polymyalgia rheumatica (pain and stiffness in the shoulder or pelvic girdle or both). Takayasu arteritis is suggested by upper extremity claudication, carotidynia (tenderness of the carotid arteries), and the absence of 1 or more pulses in a young woman.
Suspicion of WG should be raised by recurring sinusitis or otitis unresponsive to antibiotics, especially if it is associated with constitutional symptoms, epistaxis, nasal ulcerations, and nasal septum perforation. The most common presenting manifestations of CSS are worsening asthma, pulmonary infiltrates, and eosinophilia.
The possibility of vasculitis should receive strong consideration in a patient who presents with pulmonary-renal syndrome (shortness of breath, cough, hemoptysis, and pulmonary infiltrates in combination with hematuria, proteinuria, hypertension, and rapidly progressive renal failure). The antineutrophil cytoplasmic antibody (ANCA)-related vasculitides (particularly MPA and WG) are the most common causes of this potentially life-threatening presentation.
Which disorders are vasculitis mimickers?
Several conditions may mimic the vasculitides because of shared clinical, laboratory, radiographic and, sometimes, pathological features. Bacterial endocarditis, cholesterol emboli, and antiphospholipid syndrome are classic examples.
How should I investigate when I suspect vasculitis?
A complete blood cell count is one of the most useful initial laboratory tests. The presence of systemic inflammation is supported by the presence of anemia (usually normocytic but occasionally microcytic), neutrophilia, or thrombocytosis, although they are nonspecific.
The erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level are elevated in most patients who have active systemic (as opposed to limited) vasculitis. In patients with GCA, the ESR and CRP level are elevated in 95% of cases.2
A urinalysis, a creatinine test, liver function testing, and chest radiography may help clinicians assess for the presence of renal, liver, or pulmonary involvement. A cryoglobulin test, hepatitis C serology, and complement are indicated for patients who present with palpable purpura to rule out CV because this condition is related to hepatitis C infection in more than 80% of cases.3
Hepatitis B serology is indicated if PAN is suspected, although this association is becoming much rarer, now being seen in fewer then 10% of cases.4 Rheumatoid factor and elevated titers of antinuclear antibodies usually are present in cases of vasculitis resulting from an underlying connective tissue disease.
Patients in whom WG, MPA, or CSS is suspected should be tested for ANCA. The most frequently used tests are indirect immunofluorescence (IIF) assay and enzyme-linked immunosorbent assay (ELISA). The IIF test is more sensitive and ideally suited for screening.


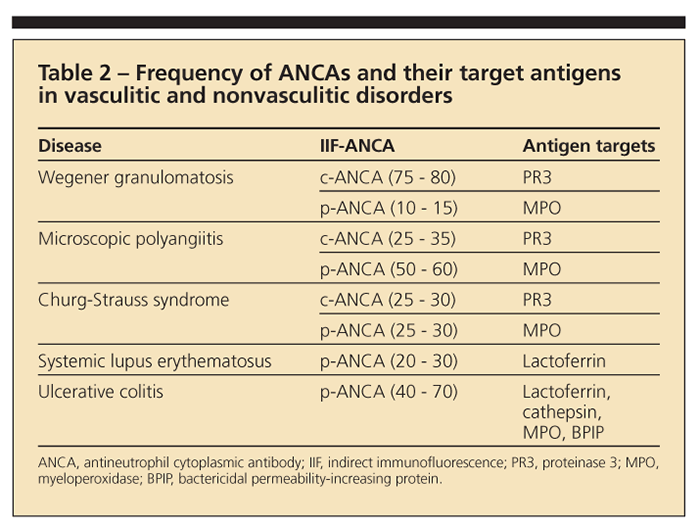
A granular cytoplasmic staining of ethanol-fixed neutrophils is typical of c-ANCA; perinuclear or nuclear staining defines p-ANCA. Proteinase 3 is the molecular target of most c-ANCAs; antigens that can cause p-ANCA reactivity include myeloperoxidase (MPO), elastase, cathepsin, and lactoferrin. Because only MPO-ANCAs have been associated with vasculitis, confirming a positive p-ANCA result with an ELISA directed specifically against MPO is important. Table 2 shows the frequency of ANCAs with IIF testing and their molecular targets in vasculitic and some nonvasculitic conditions.5
CT scans of the sinuses and lungs are much more sensitive than plain x-ray films in patients who have sinus and lung involvement resulting from WG. CT and MR angiography are the imaging modalities of choice for patients with large-vessel vasculitides.
What about tissue biopsy?
Tissue biopsy remains the best way to confirm a diagnosis of vasculitis. When the skin is affected, it is the most easily accessible tissue. Fresh lesions (less than 24 hours) should be selected; specimens should be sent for both light microscopy and immunofluorescence, especially when Henoch-Schnlein purpura (which is characterized by IgA deposition within the vessel walls) is being considered.
We recommend a temporal artery biopsy in all patients in whom GCA is suspected. The presence of temporal artery swelling or tenderness or both on physical examination increases the yield of a positive biopsy result. The false-negative result rate of a unilateral biopsy ranges between 20% and 40%; performing biopsies on both sides reduces this rate to less than 10%.6 Color Doppler ultrasonography performed by practitioners experienced with the technique eventually may replace the need for biopsy, especially in patients who have high and low pretest probability.
Biopsies of the nasal mucosa often are performed in patients in whom WG is suspected, but their diagnostic yield is very low (20% to 30%) because of the small amount of tissue obtained. The yield of transbronchial or needle biopsies of lung lesions is even lower (less than 10%), and open lung biopsies are necessary to confirm the diagnosis of pulmonary vasculitis (90% yield).7 Sural nerve biopsies are diagnostic in only 40% to 50% of patients who are symptomatic, but combining them with a gastrocnemius biopsy increases the yield significantly.In patients who have an active urinary sediment, renal biopsies rarely show vasculitis per se; instead, they show a pauci-immune necrotizing glomerulonephritis, which is diagnostic of ANCA-related vasculitides. In contrast, patients with PAN may have renal infarcts resulting from occlusion of medium-size vessels. Renal biopsy can be dangerous in these patients and should be avoided. Abdominal angiography is the procedure of choice because it may show areas of long segments of stenosis alternating with dilations and microaneurysms, features highly supportive of the diagnosis of medium-vessel vasculitides.
Can ANCA testing replace biopsy?
As noted previously, ANCA tests are sensitive and very specific for the diagnosis of active WG or MPA. Therefore, in clinical situations in which the pretest probability is moderate to high (50% to 80%), we think that a positive ANCA result with both IIF and ELISA testing can replace the need for an invasive tissue biopsy to confirm the diagnosis.
How should I treat my patients who have vasculitis?
It depends on the extent of disease. Patients who present with palpable purpura and have no history of recent drug exposure, negative cryoglobulins and hepatitis serological test results, and no clinical or laboratory evidence of other organ involvement (idiopathic cutaneous vasculitis) may require no treatment other than reassurance and perhaps an NSAID or, if the rash is extensive, low-dose prednisone (less than 0.5 mg/kg) for a few days.
For patients who have recurring episodes of idiopathic CV, 0.6 mg of colchicine 2 or 3 times a day might be considered. If this treatment is ineffective, 50 to 200 mg/d of dapsone is a good choice as long as the patient's blood work is monitored for drops in hemoglobin level because this medication can cause hemolysis, particularly in patients with a glucose-6-phosphate dehydrogenase deficiency.
What about giant cell arteritis?
Corticosteroids remain the cornerstone of treatment for patients with GCA. Because histological changes sufficient to confirm the diagnosis will remain up to 1 month after treatment is started, therapy should not be delayed until the biopsy is performed.
For patients who present with vision loss in 1 eye or who have symptoms suggestive of impending vision loss (eg, diplopia and amaurosis), we use pulse methylprednisolone, 500 to 1000 mg/d IV for 3 days, followed by prednisone, 1 mg/kg up to a maximum of 60 mg/d. In the absence of vision symptoms, we do not use pulse methylprednisolone; therapy with oral prednisone is started at the same dosage as above.
Symptoms typically disappear within days, and acute phase reactants (ESR and CRP level) normalize within a few weeks. Our patients continue to receive the same prednisone dosage for 2 to 4 weeks before it is tapered-by 10 mg every 1 to 2 weeks down to 40 mg, 5 mg every 1 to 2 weeks down to 20 mg, 2.5 mg every 1 to 2 weeks down to 10 mg, and then 1 mg every month until cessation. The role of low-dose aspirin (81 to 325 mg/d) in reducing the risk of cranial ischemic complications, particularly during the first month of therapy, is controversial.
Proper measures should be taken in all patients to reduce the risk of corticosteroid-induced osteoporosis, including the use of bisphosphonates, calcium, and vitamin D supplementation. Regular monitoring of blood sugar levels and blood pressure also is important. We suggest monitoring the patient's ESR or CRP level on a monthly basis, but we advise against changing the tapering schedule only on the basis of an asymptomatic elevation of acute phase reactant levels.
Adjunctive treatment with methotrexate (MTX) may lower the risk of relapse and reduce the cumulative dose of corticosteroids.8 However, using infliximab as maintenance therapy in patients with GCA appears to be of no benefit and even may be harmful.9
What about Wegener granulomatosis?
Cyclophosphamide (CYC) in combination with corticosteroids remains the treatment of choice for patients with life- or organ-threatening manifestations of WG or MPA. This combination is successful in inducing remission in at least 85% of patients within 9 months (median time to remission, about 3 months).10
CYC may be given orally (2 mg/kg/d in patients younger than 75 years who have normal renal function) or by intravenous pulses (15 mg/kg up to 1200 mg every 2 weeks × 3 doses and then every 3 weeks until remission).11 With the oral route, we give the total daily dosage in the morning and advise our patients to drink at least 3 tall glasses of water or juice to facilitate rapid excretion of the drug and prevent hemorrhagic cystitis.
Prolonged administration of this drug is associated with an increased risk of bladder cancer. We recommend long-term monitoring with quarterly urine cytology for all patients who have been exposed to the drug for longer than 1 year.
The risk of infertility, another important problem for both sexes, is related to the dosage and duration of treatment. The age of women at the time of drug administration is important; older women are at higher risk for infertility than younger women.
With either route of administration, we routinely prescribe trimethoprim/sulfamethoxazole (TMP/SMX), 1 double-strength tablet 3 times a week, to prevent Pneumocystis carinii(jiroveci) pneumonia. Once remission is achieved (usually within 3 to 6 months), we stop the CYC and switch to azathioprine, 2 mg/kg/d, or MTX, 0.3 mg/kg/wk up to 25 mg/wk.10,12 The duration of treatment with these agents once remission has been achieved remains a subject of controversy among vasculitis experts. However, most agree with continuing treatment for a minimum of 18 months.
The corticosteroid regimen is similar to the one used for patients with GCA. However, on the basis of recent evidence suggesting that patients for whom prednisone is discontinued tend to relapse more frequently than those who continue to receive the medication (42% vs 13% during a mean follow-up of 22 months), we tend to continue treatment at 5 mg to 7.5 mg for a minimum of 1 to 2 years.13
A trial performed in patients with non–life-threatening manifestations demonstrated similar remission rates in patients treated with MTX and those treated with CYC.14 However, the relapse rate was higher in MTX-treated patients. Some evidence supports the use of 1 double-strength tablet of TMP/SMX twice daily to reduce upper respiratory tract disease flares.15 Etanercept is not effective for the maintenance of remission in WG and is associated with a high rate of treatment-related complications.16 In contrast, rituximab recently was shown to induce remission at a rate similar to that of cyclophosphamide in those patients with severe ANCA-related vasculitides.17
References:
References1. Stone JH, Nousari HC. “Essential” cutaneous vasculitis: what every rheumatologist should know about vasculitis of the skin. Curr Opin Rheumatol. 2001;13:23-34.
2. Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurrence in a population-based study. Arthritis Rheum. 2001;45:140-145.
3. Vassilopoulos D, Calabrese LH. Hepatitis C virus infection and vasculitis: implications of antiviral and immunosuppressive therapies. Arthritis Rheum. 2002;46:585-597.
4. Guillevin L, Mahr A, Collard P, et al; French Vasculitis Study Group. Hepatitis B virus-associated polyarteritis nodosa: clinical characteristics, outcome, and impact of treatment in 115 patients. Medicine (Baltimore). 2005;84:313-322.
5. Wiik AS. Clinical use of serological tests for antineutrophil cytoplasmic antibodies: what do the studies say? Rheum Dis Clin North Am. 2001;27:799-813.
6. Hall S, Persellin S, Lie JT, et al. The therapeutic impact of temporal artery biopsy. Lancet. 1983;26:1217-1220.
7. Duna GF, Galperin C, Hoffman GS. Wegener's granulomatosis. Rheum Dis Clin North Am. 1995;21:949-986.
8. Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum. 2007;56:2789-2797.
9. Hoffman GS, Cid MC, Rendt-Zagar KE, et al; Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. 2007;146:621-630.
10. Jayne D, Rasmussen N, Andrassy K, et al; European Vasculitis Study Group. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med. 2002;349:36-44.
11. de Groot K, Harper L, Jayne DR, et al; EUVAS (European Vasculitis Study Group). Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009;150:670-680.
12. Pagnoux C, Mahr A, Hamidou MA, et al; French Vasculitis Study Group. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med. 2008;359:2790-2803.
13. Walsh M, Merkel PA, Mahr AD, Jayne D. The effect of low-dose corticosteroids on risk of relapse in ANCA-associated vasculitis: a systematic review and meta-analysis of clinical trials. Arthritis Rheum. 2007;56:S766.
14. De Groot K, Rasmussen N, Bacon PA, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2005;52:2461-2469.
15. Stegeman CA, Tervaert JW, de Jong PE, Kallenberg CG. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener's granulomatosis. Dutch Co-Trimoxazole Wegener Study Group. N Engl J Med. 1996;335:16-20.
16. Wegener's Granulomatosis Etanercept Trial (WGET) Research Group. Etanercept plus standard therapy for Wegener's granulomatosis. N Engl J Med. 2005;352:351-361.
17. Stone JH, Merkel PA, Seo P, et al. Rituximab versus cyclophosphamide for induction of remission in ANCA-associated vasculitis: a randomized controlled trial (RAVE). Arthritis Rheum. 2009;60:S204.
















